准备文件:关联出的相关位点、gff注释文件
1、确定区间:
查看文献以及参考bedtools使用的软件流程,确定显著位点上下游10kb的基因。
注:10kb可以根据自己的要求进行修改
# LeafS_new_GLM_GWAS7.bed文件是关联出的位点,合并位点信息,生成qujian文件
bedtools merge -i LeafS_new_GLM_GWAS7.bed >snp_qujian.bed2、提取gff中染色体信息
#使用全部的gff文件会报错,位点信息都在4号染色体上,于是提取4号染色体的gff文件
grep '^chr04' t2.gff3 > chr04_records.gff3
#将提取出的染色体进行排序
bedtools sort -chrThenSizeA -i chr04_records.gff3 >chr04_size.gff33、筛选候选基因
bedtools intersect -a chr04_size.gff3 -b snp_qujian.bed -wa >pvalue7_chr04_10kb_gene.txt
#pvalue7_chr04_10kb_gene.txt是注释的区间,从中挑选基因信息和其他信息4、NCBI上比对
选择几个近缘物种
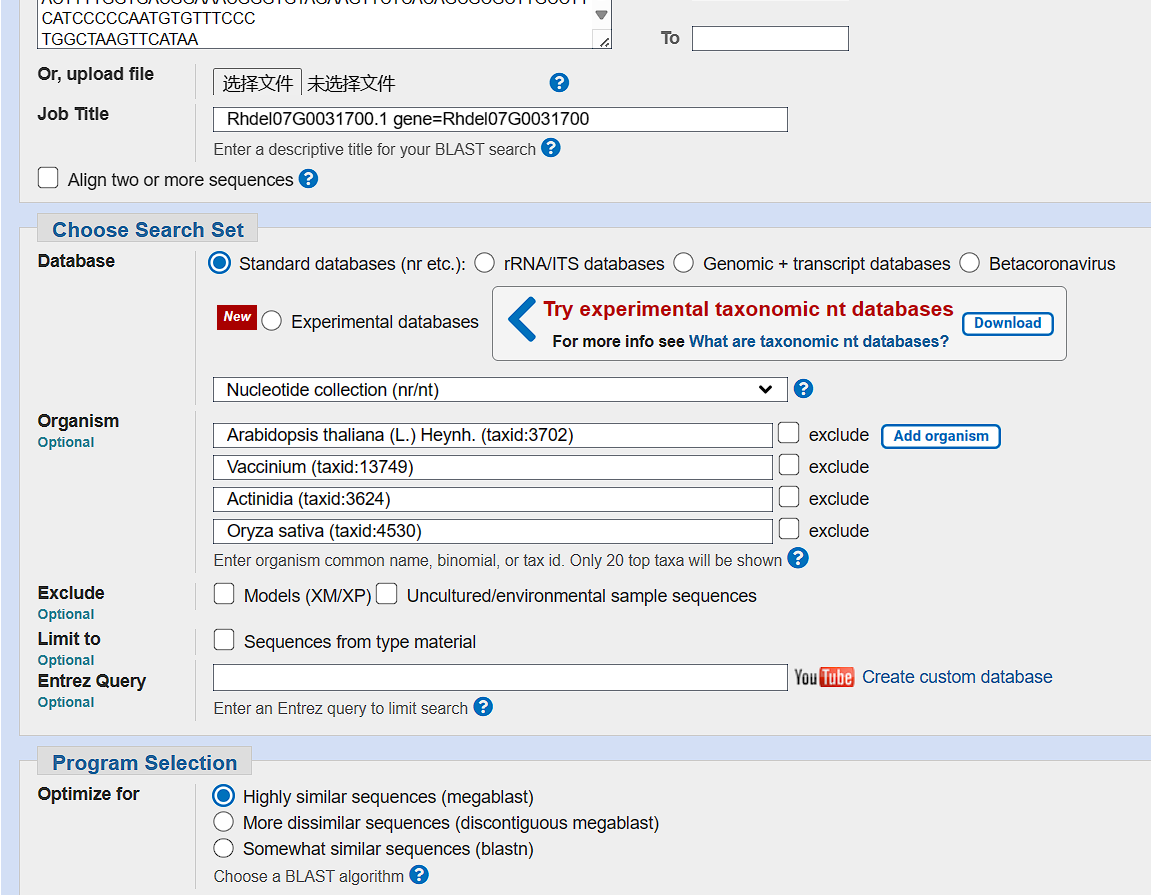
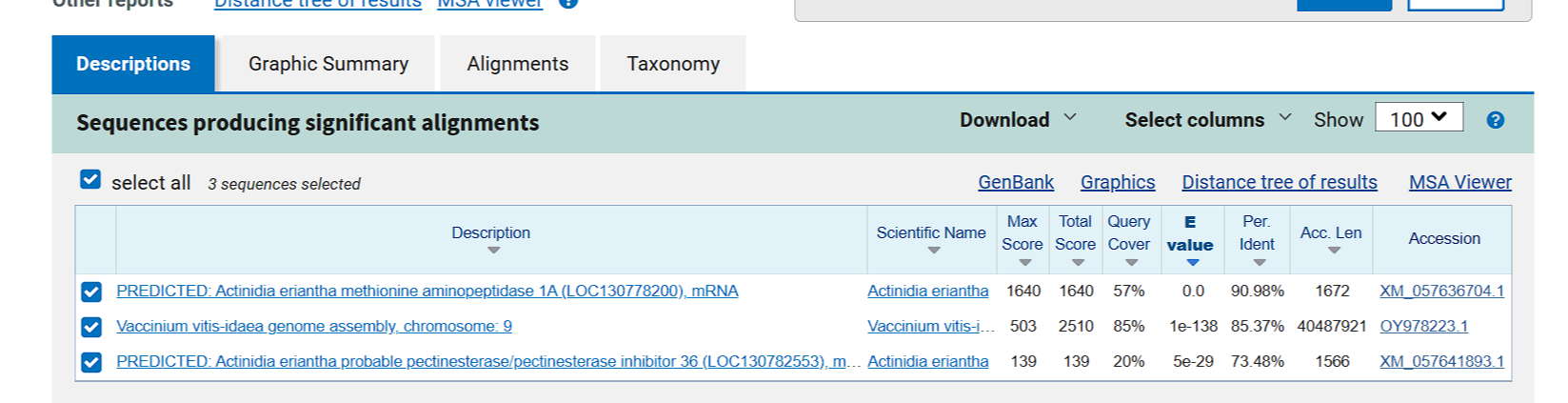
结果如下:
参考:使用bedtools进行gwas基因注释_如何研究没有注释的基因-CSDN博客