单一模型的GWAS结果过拟合并不意味着该表型不受SNP效应的影响
需要将可用的模型全部测试之后才能确认。BLINK和GLM通常用于测试,但其他模型也需要进行分析后才能得到结论
(1)直接计算x和y的相关性:一般线性模型(GLM)
由于直接进行回归分析,不涉及其他条件,可能存在较多的假阳性问题。

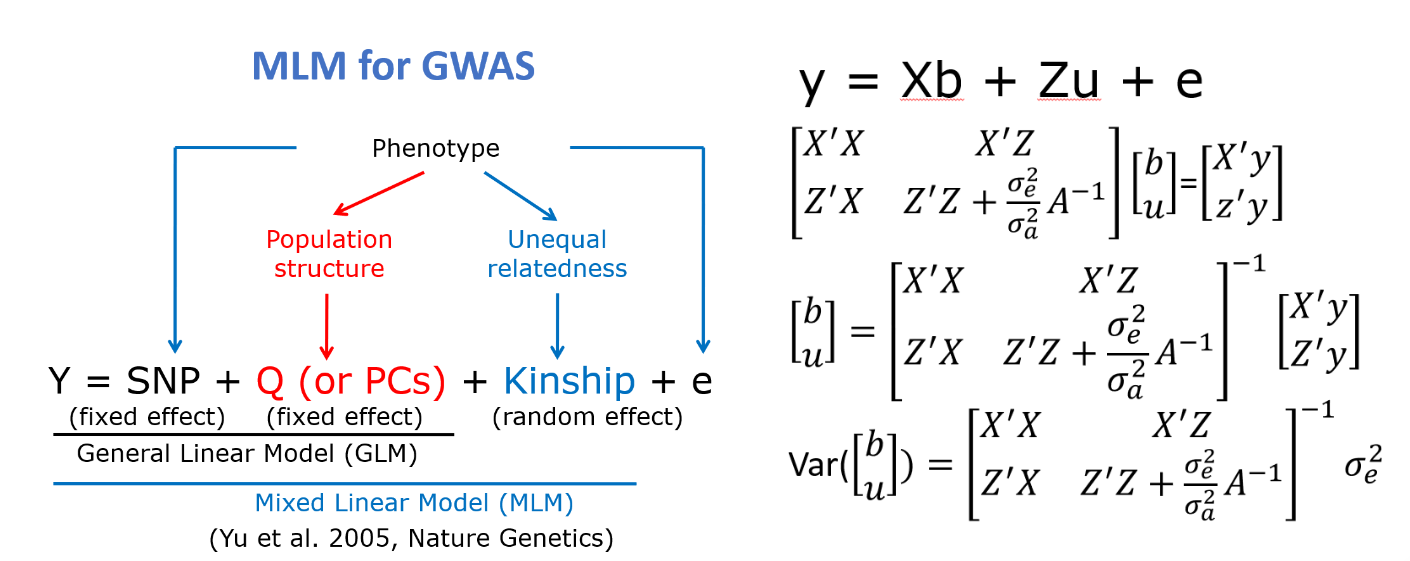
(2)矫正假阳性:混合线性模型(MLM)
在模型中插入了亲缘关系矩阵,剔除群体本身的遗传差异以得到更准确的结果。
(3)降低过滤阈值:压缩混合线性模型(CMLM)
MLM矫正过于严格,一些实际相关的SNP标记也会被过滤掉,因此最好的选择是直接找到那些假阴性的SNP标记并将其剔除掉。(倒洗澡水的时候把孩子也倒掉了)将个体按照亲缘关系的相似性进行分组,组内个体的亲缘关系都是一样的。根据个体遗传关系的相似性,把大量的个体压缩到数个组,然后用组当做协变量,消除了部分数亲缘关系的协变量影响。
(4)SUPER(Settlement of Kinship Under Progressively Exclusive Relationship, 逐步排他性亲缘关系解决方案)
重新计算亲缘关系矩阵,使用跟表型相关的所有SNP,但是排除掉测试用的SNP来重新构建亲缘关系矩阵。
(5)FarmCPU(Fixed And Random Model Circuitous Probability Unification)
首先把随机效应模型的亲缘关系矩阵(kinship)转换为固定效应的关联SNP矩阵(QTN)以提升计算速度。其次利用QTN矩阵当做协变量时,重新做关联分析,准确率也会提升。
(6)Blink
在SUPER和FarmCPU应用的时候,有一个关键的点是把bin(一段区域,比如10kb)当做SNP单位,而不是单个的SNP。这就好造成一个问题,因为基因在染色体上不是均匀分布的,LD(连锁不平衡距离)在不同位置也不一样大。所以根据实际染色体位置选择对应的bin大小是更加可行的策略。
如上面的实例图,在进行GWAS分析时候,先用上方的GLM模型获得QTNs,然后用右侧的GLM以QTNs当做协变量进行SNP检测,得到的SNP根据LD信息确定QTNs的信息,进而利用左侧的GLM以BIC(Bayesian information criterion)策略进行QTNs准确性检测,排除假设错误的部分,保留真实的QTNs,这是一个循环,然后重复不断进行,直到检测到所有关联SNP(也就是QTNs)。