#了解plink文件
(1)map文件
map文件有4列:染色体, SNP名称, SNP位置, 碱基对坐标
SNP位置通常没有,可以使用0代替,也可以用-9表示(-9在plink中代表缺失)
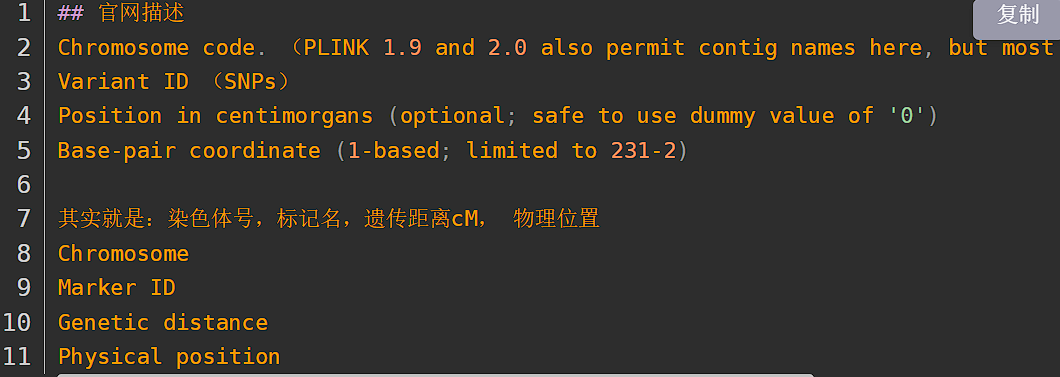
例子:
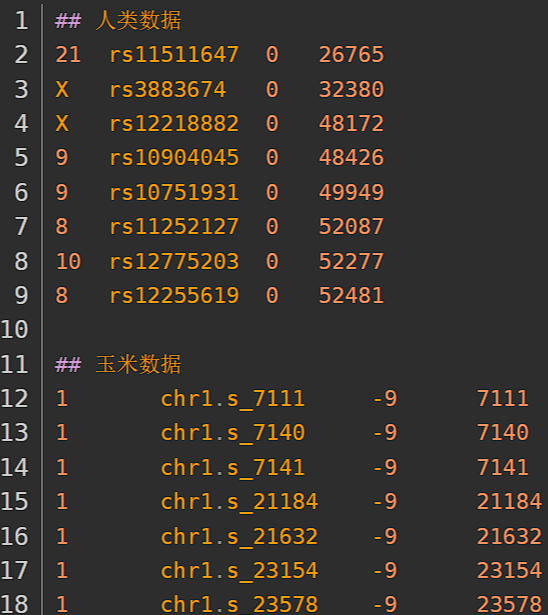
杜鹃花数据:
(2)ped文件
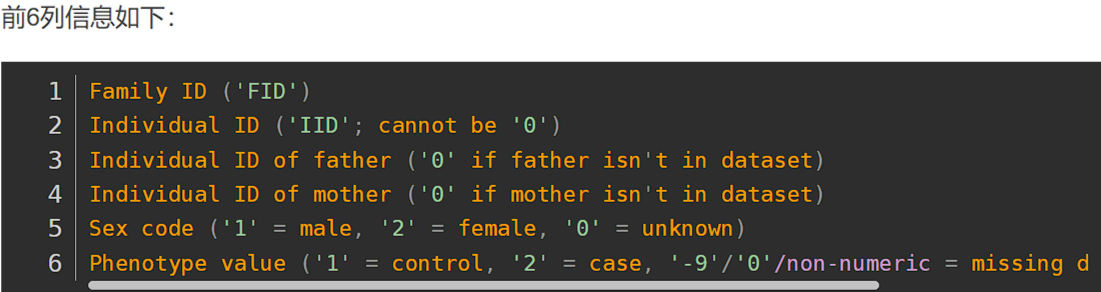
主要包括SNP的信息, 包括个体ID, 系谱信息, 表型和SNP的分型信息。
Ped文件没有表头,每行包含6+2V个数据(空格或tab分割),前6列为系谱信息列,其中2V为基因型列。第7列开始为基因型列。
其中第7列和第8列为第一个材料的基因型,第9列和第10列为第二个材料的基因型,以此类推,因此,V个材料有2V列表示其基因型。
例子:
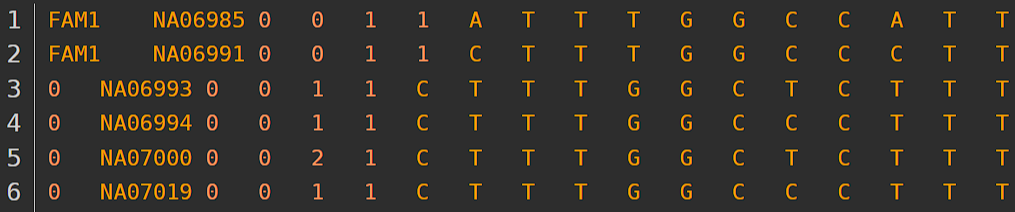
杜鹃花数据:

(3)fam文件
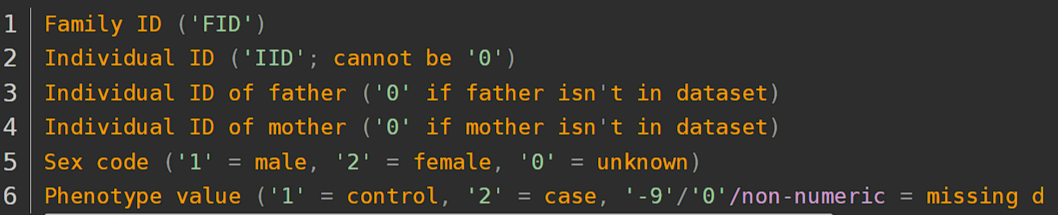
例子:
杜鹃花的数据:
(4)bim文件
例子:
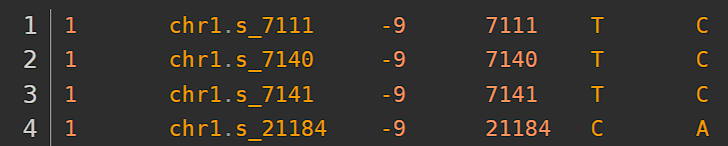
杜鹃花数据:
#质控:
缺失率、最小等位基因频率(MAF)、哈迪温伯格平衡检验(HWE)等等
数据筛选几个标准:
1、去除缺失率大于10%的SNP位点
2、样本检出率大于90%
3、最小等位基因频率(MAF)大于1%
4、去除哈迪温伯格平衡检验(HWE)P值小于10的-6次方的SNP
缺失率、最小等位基因频率(MAF)、哈迪温伯格平衡检验(HWE)
#目前主要使用的是:
LD 计算两个SNP位点的连锁不平衡值: --ld
plink --vcf combine_all.pass.recode.clear.vcf --allow-no-sex --maf 0.05 --geno 0.2 --r2 --ld-window 999999 --ld-window-r2 0.5 --allow-extra-chr --out combine_all.pass.recode.clear.0.5.vcf例子:
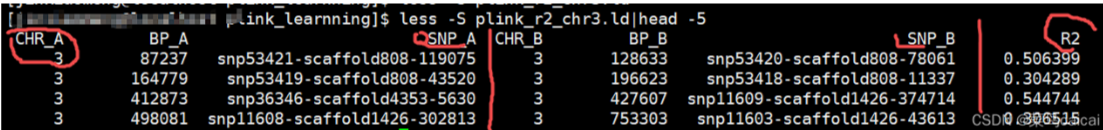
杜鹃花数据:
生成文件后,将ld文件的内容删除
#pca分析:使用 -pca
–pca 参数用于对数据做pca分析,后面的10是取前十个pca结果的意思,一般不需要取太多,因为最后是用最显著的第一列和第二列作图。
–threads是为了增加线程,
–out参数后面跟输出文件名,可以自己设定。
最终得到两个文件:.eigenval 和.eigenvec,我们用.eigenvec文件进行绘图。
#文件格式转换:正常格式转换为二进制,二进制格式转换为正常格式。
总之,plink工具很强大。