以下使用香榧的TPS家族进行分型示范。软件用到的是【Tbtools】和【MEGA】。
一、准备香榧的蛋白质序列
这里使用的是ba版的蛋白质文件,路径为/data2/liyupeng/alice/output/female_anno/rename_fa_gff3/extract/female_mark1_protein.fa
二、p450基因家族的鉴定
基于 HMMER 的隐马尔可夫模型搜索
1、找到TPS家族的 HMM 模型
(1)先去interpro官网下载隐马可夫模型的全部模型数据加载下来Pfam-A models。网址:https://www.ebi.ac.uk/interpro/download/Pfam/
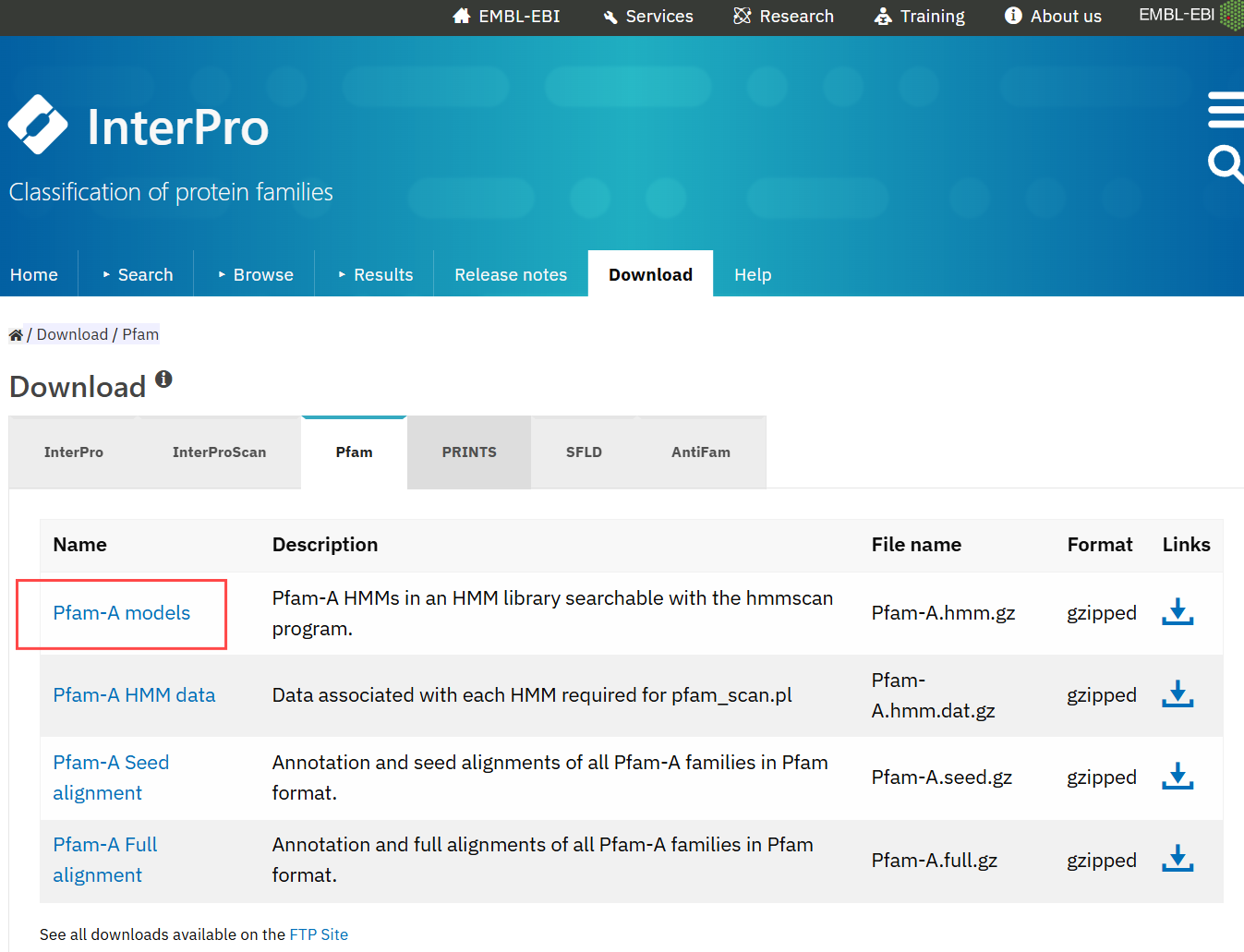
(2)从interpro数据库中获取 TPS家族的 HMM 模型(PF01397、PF03936、PF05860)。
在interpro数据库中的search > bytext中输入TPS,在结果中找到pfam号,要注意筛选。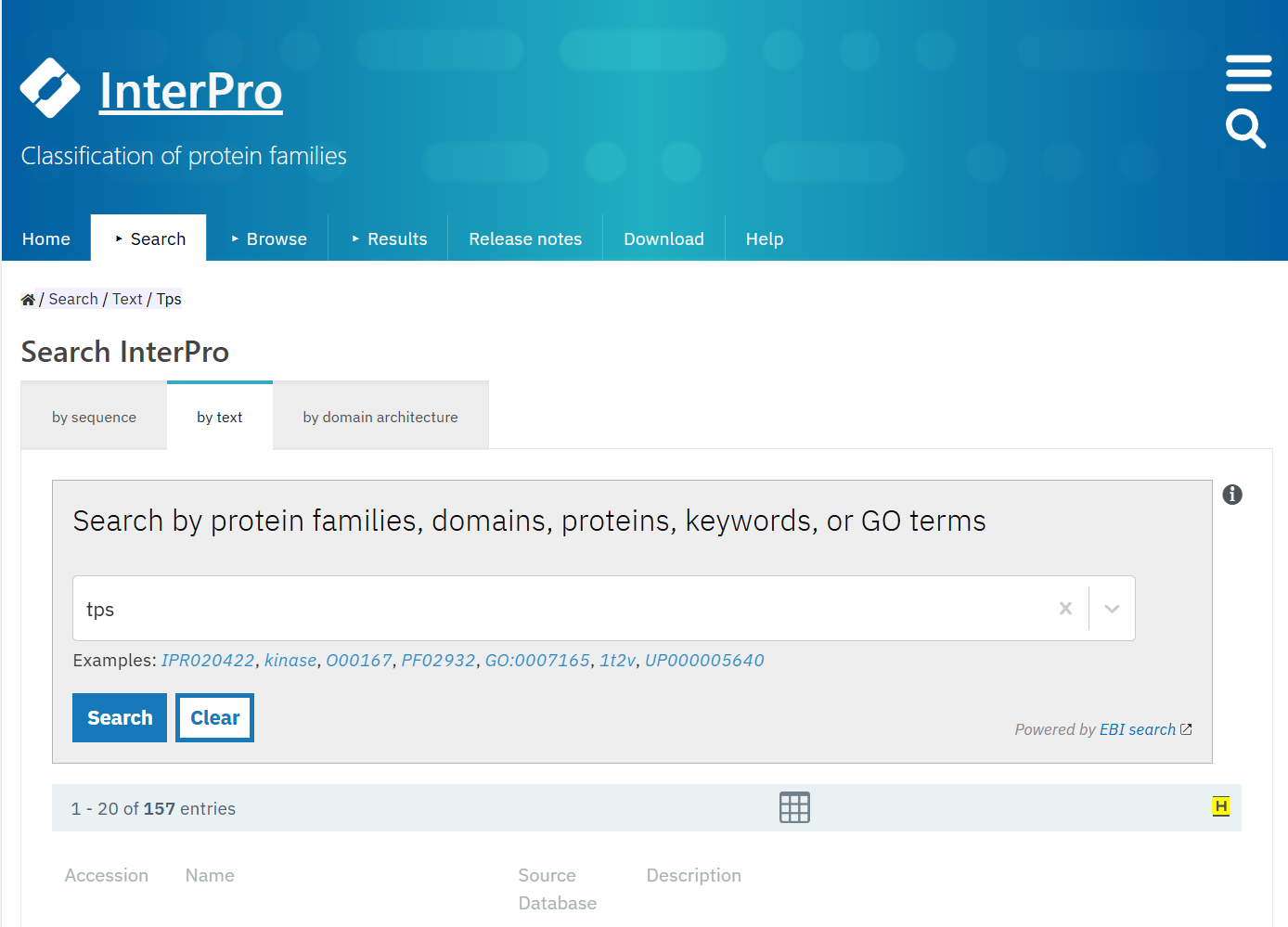
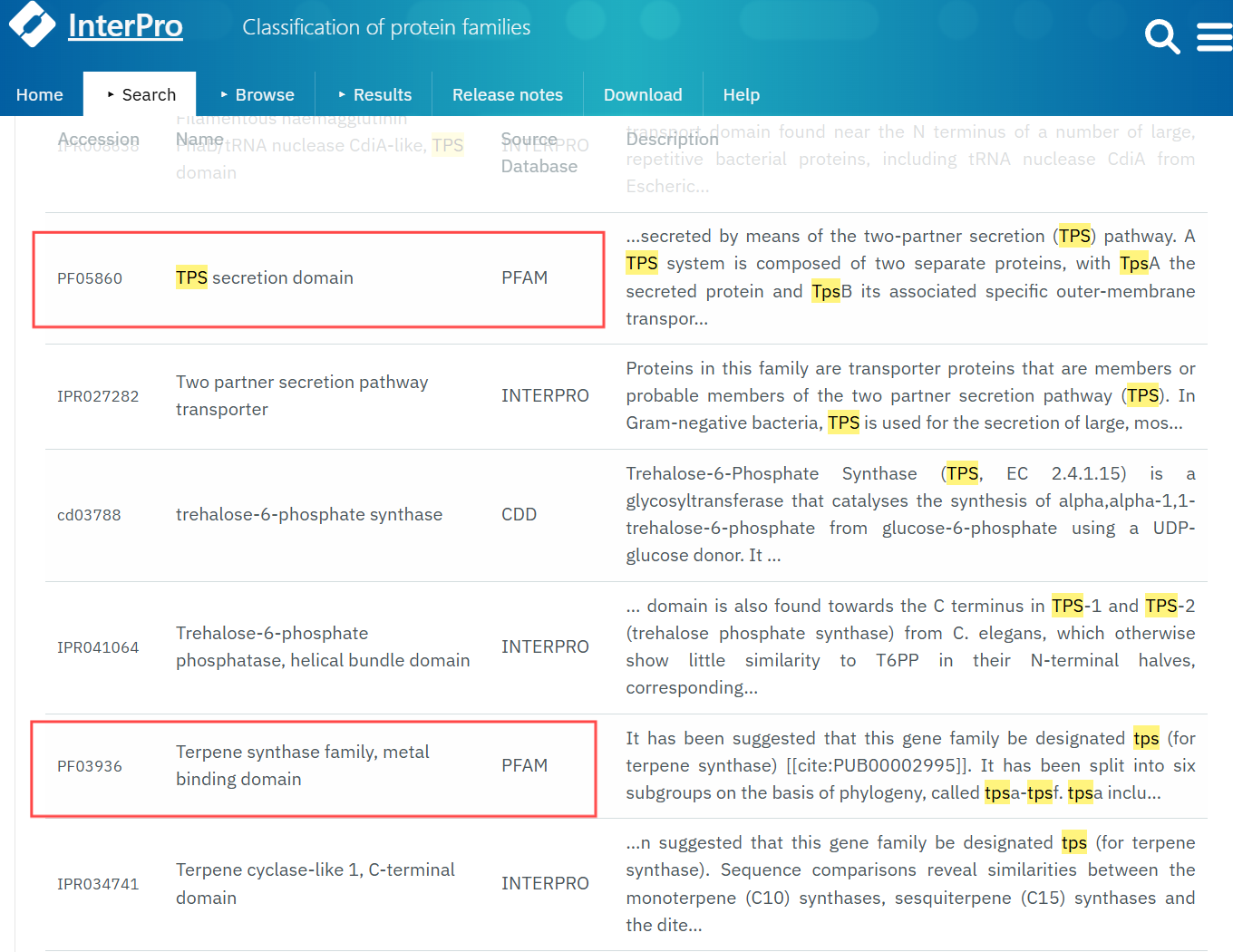
(3)找到pfam号之后,新建一个pfamid.txt,把PF00067,输入进入。
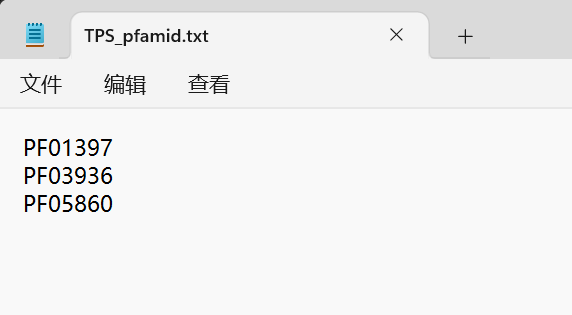
2、打开TBtools,在others > Simple HMM Search中根据对应的要求,输入蛋白质文件和pfam号,点击start,成功会有congratulations,得到TBtools.SimpleHMM.Result.xls文件。
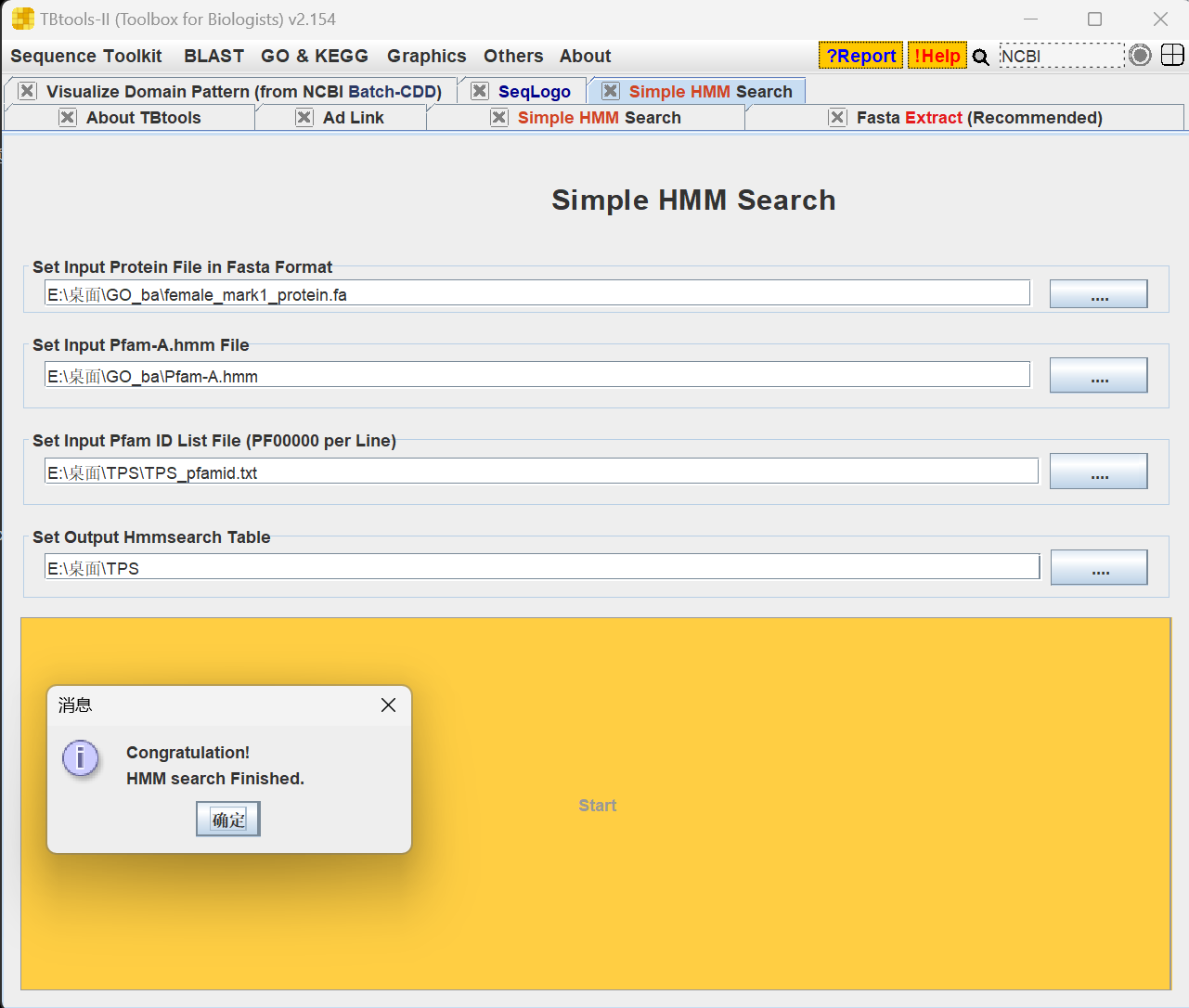
(1)TBtools.SimpleHMM.Result.xls文件里面有两个表,# Sequence scores(全长的打分)和# Domain scores(保守结构域区域的打分),我们只需要第一个表的id。
由于表格中的所有数据都在一列中,可以对表格进行分列处理,得到第一列的id号。可以选定一个标准。evalue<e的负10次方来筛选,xlxs表里面是4.54e-05。n的意思就是有几个结构域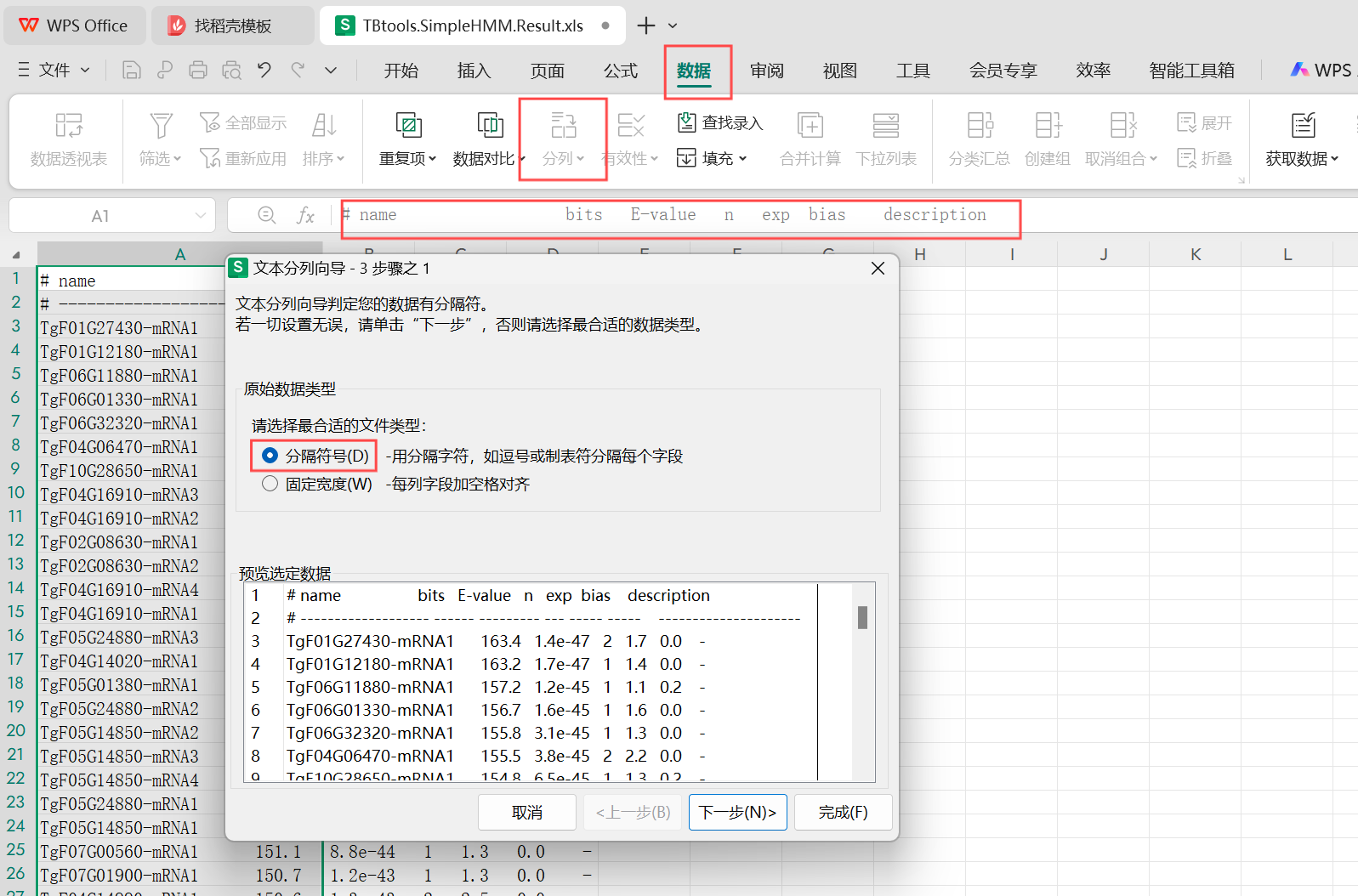
获得了id后之后我们再通过蛋白质文件把这些蛋白质序列提取出来进行多序列比对和进化树分析。
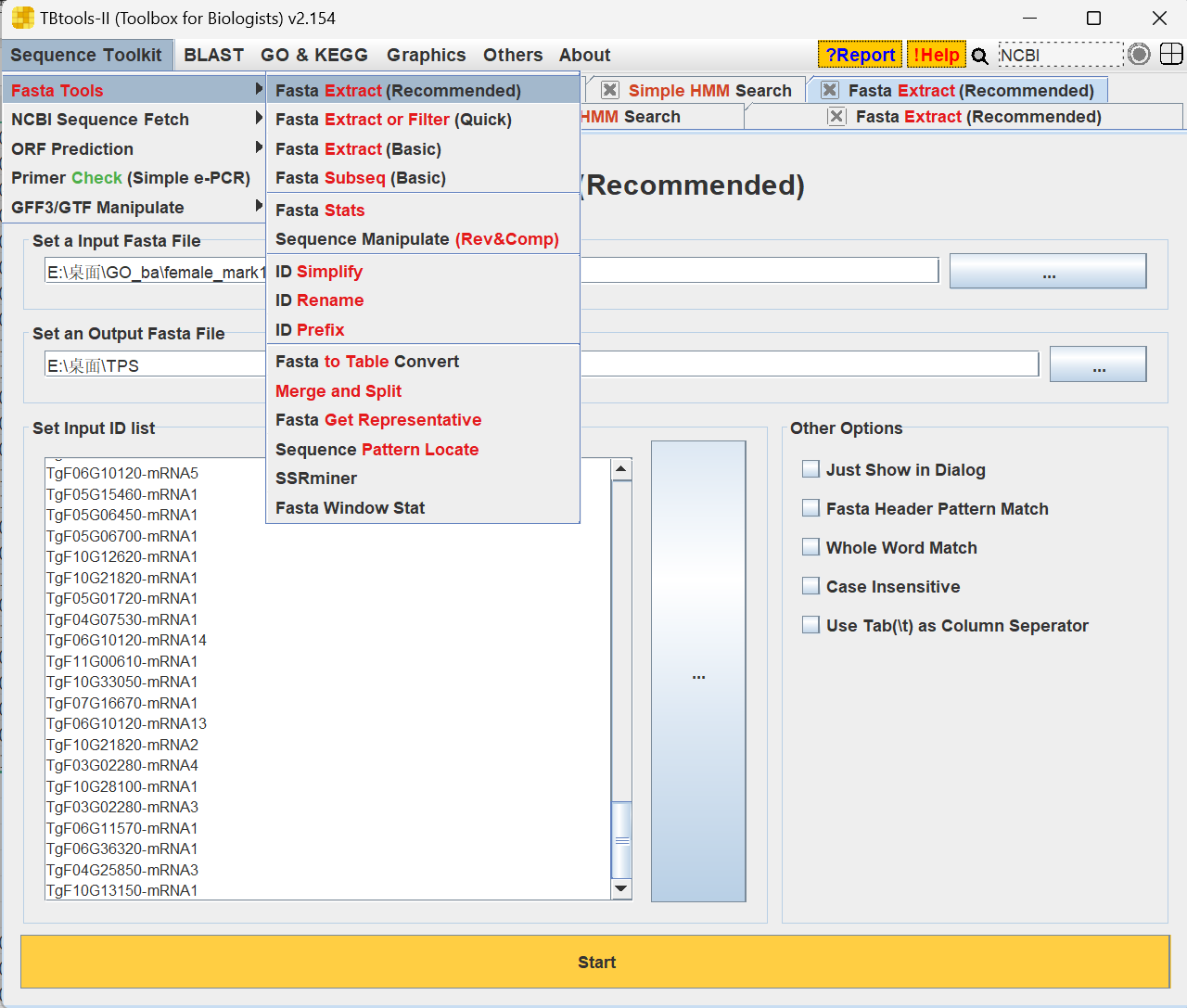
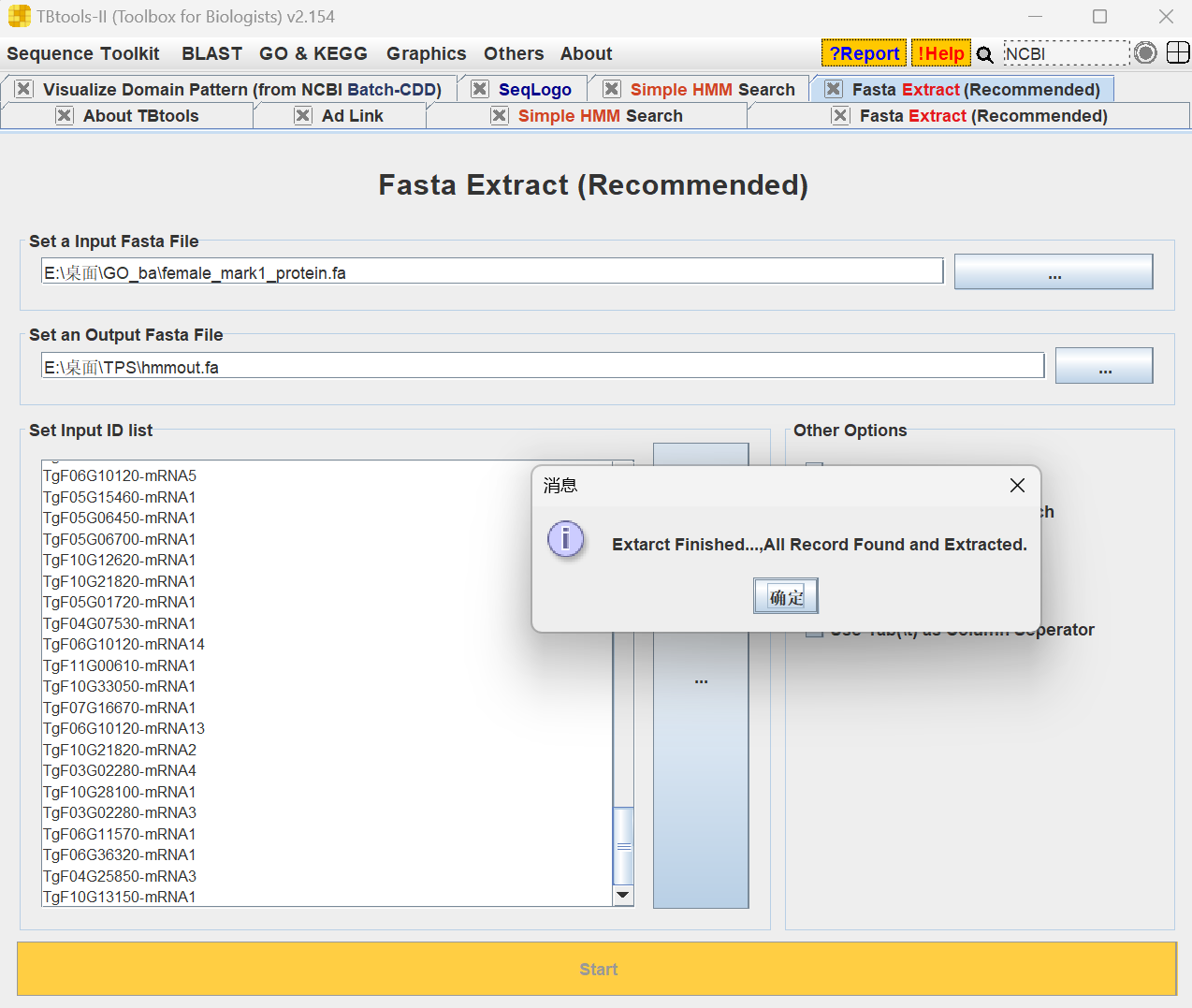
(2)打开TBtools > SequenceToolkit > Fasta Tools > Fasta Extract(Recommended),把上面获取的id输入进去,香榧的蛋白质文件输入对应位置,指定输出位置以及输出文件名hmmout.fa,点击start,把id对应的蛋白序列提取出来。
使用ncbi的cdd—search去验证hmm鉴定出来的基因家族是不是真的全部都含有这个结构域,用序列比对的方式去验证,如果有假阳性的结果就删除掉。
1、打开ncbi的cdd—search,输入上一步得到的 hmmout.fa文件,得到文件hitdata.txt。网址:https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi。
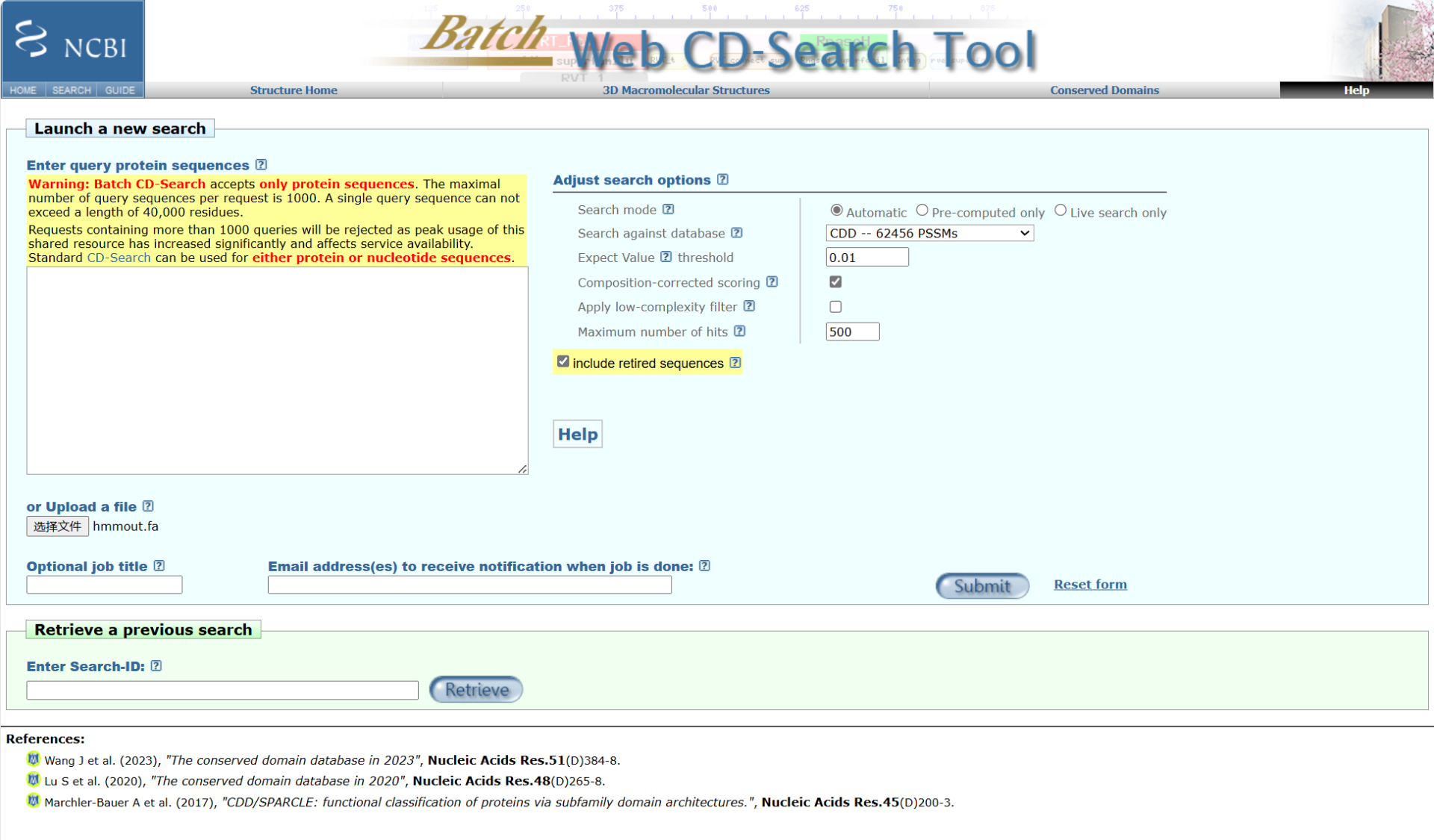
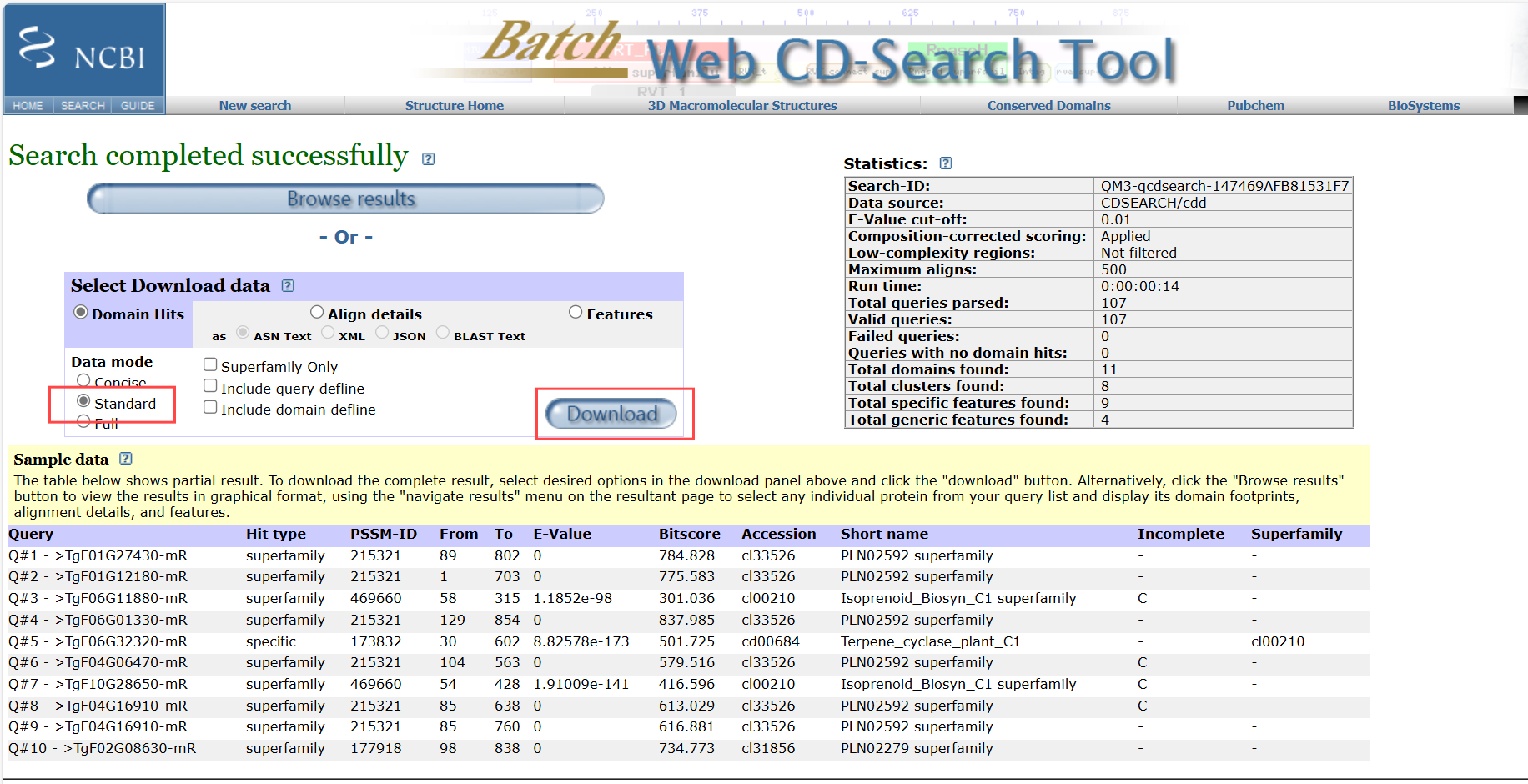
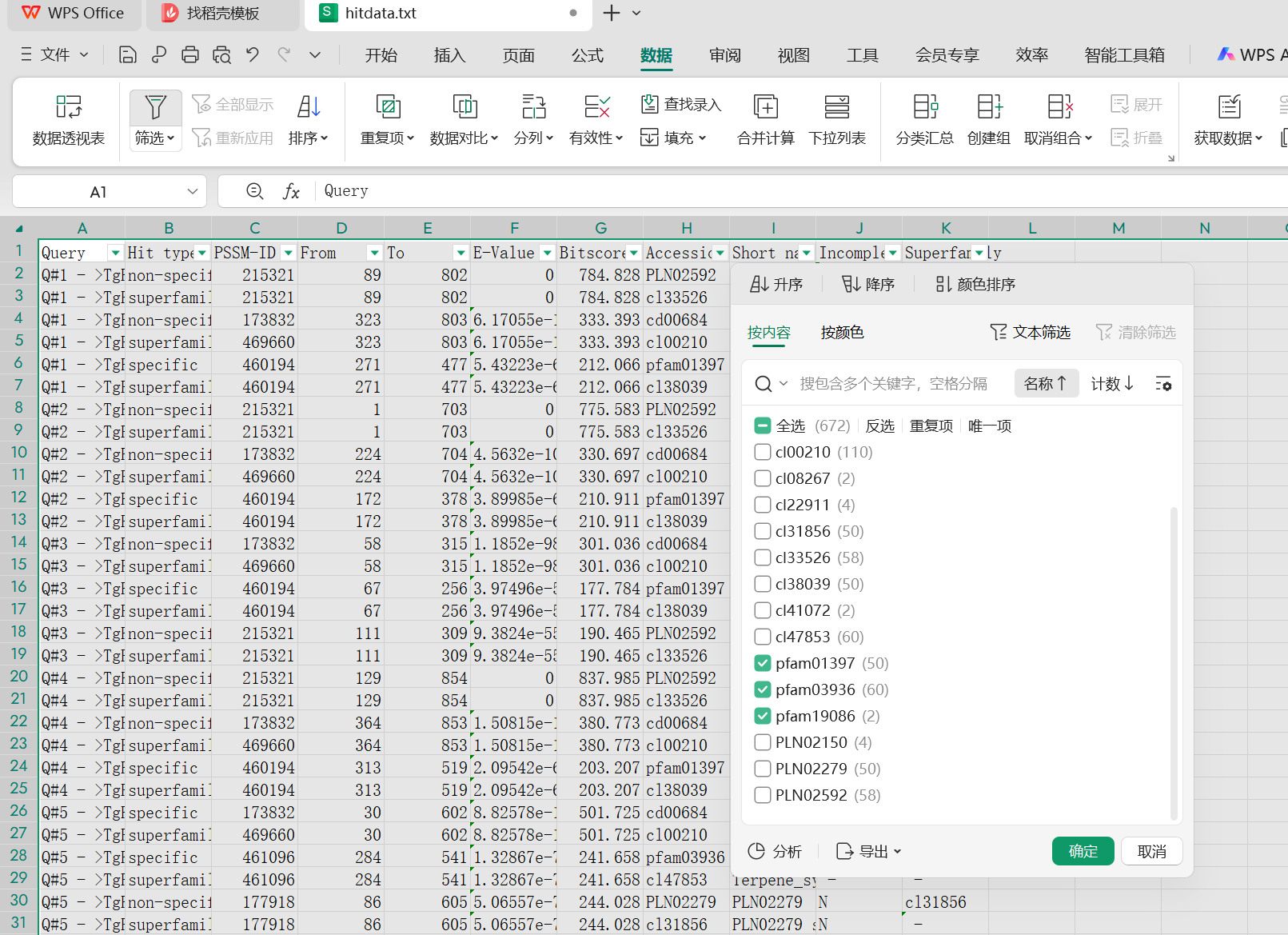
2、打开hitdata.txt文件删除前面注释行,Query就是我们要查询的序列。我们需要筛选包含我们目标的结构域的蛋白数量。和我们hmm鉴定出来的数量进行比对,变少说明我们hmm鉴定的数量有些是假阳性,需要删除。
(1)通过表格筛选Accession,选择pfam号。在这里拉到最后可以看到有107个id,我们通过hmm模型鉴定的id也是107个。可以直接用这个id