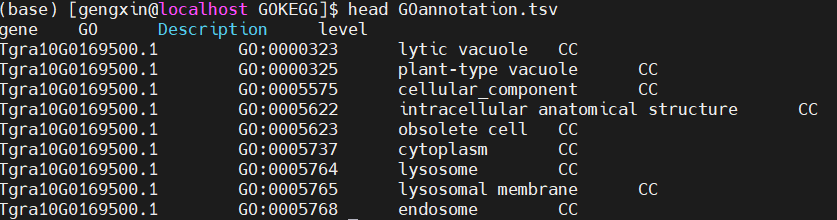
使用到的软件是R和Rstudio,在官网可以下载
一、使用eggNOG对基因组进行注释
分析需要的网站:eggnog-mapper 网址:http://eggnog-mapper.embl.de/
提交蛋白的fasta文件并填写邮箱地址
PS:这里需要注意在Annotation options设置中对Taxonomic Scope进行选择,选择Viridiplantae-33090,将其注释到植物通路上面
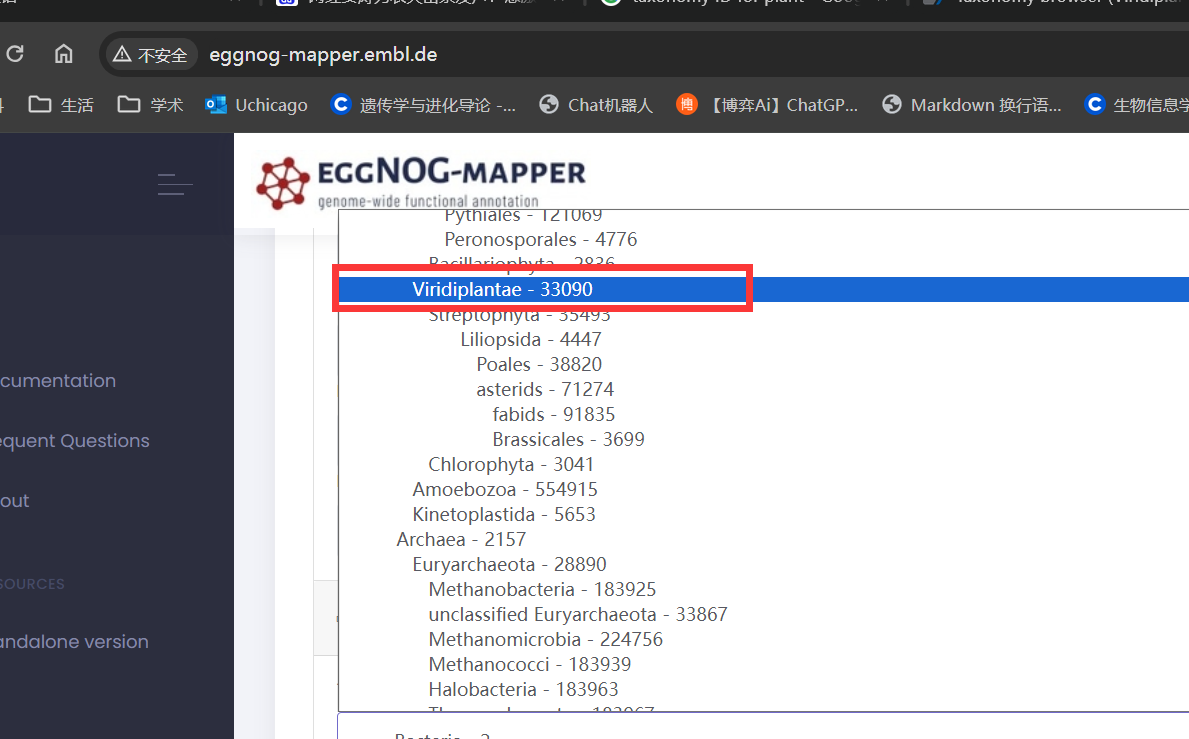
之后去邮箱里验证就能跳转过去start job了
之后等待结果,结束以后会发邮件然后点击Access your job files here尽快下载结果即可
得到一个名为out.annotation.csv格式文件将#号注释的行和query前的#删掉即可
二、解析eggNOG文件
1、首先需要去GO下载GO的obo文件,这里使用go-basic.obo然后用parse_go_obofile.py脚本可以把obo文件解析为如下格式:
python parse_go_obofile.py -i go-basic.obo -o go.tb处理后的文件为
[root@localhost go_ba]# head go.tb
GO Description level
GO:0000001 mitochondrion inheritance BP
GO:0000002 mitochondrial genome maintenance BP
GO:0019952 reproduction BP
GO:0050876 reproduction BP
GO:0000003 reproduction BP
GO:0000005 obsolete ribosomal chaperone activity MF
GO:0000006 high-affinity zinc transmembrane transporter activity MF
GO:0000007 low-affinity zinc ion transmembrane transporter activity MF
GO:0000013 obsolete thioredoxin MF2、eggNOG注释完后,下载注释结果数据,使用另一个parse_eggNOG.py脚本处理eggNOG的结果
python parse_eggNOG.py -i panax_ginseng.annotations -g go.tb -O ath,osa -o ./「参数说明」
「-i」 eggNOG的注释结果
「-g」 上一步根据obo解析出来的文件
「-O」 参考物种(只用于KEGG注释,使用KEGG三字母物种缩写表示).设置这个参数的原因是我做KEGG富集的时候发现有的基因会出现在非常荒唐的通路上,比如某个植物基因富集到了癌症的相关通路,后来发现原因是有的比较基础的KO可能与癌症通路有关,如果不使用参考物种,直接用KO去寻找map的话就会出现上述的情况。这里使用参考物种可以把没有出现在参考物种中的通路给过滤掉。植物选择拟南芥和水稻作为参考。
「-o」 输出结果文件夹。会在该文件夹生成GOannotation.tsv和KOannotation.tsv两个文件
parse_go_obofile.py parse_eggNOG.py脚本路径:92服务器/home/gengxin/TG_annotation
处理后的GO和KEGG注释文件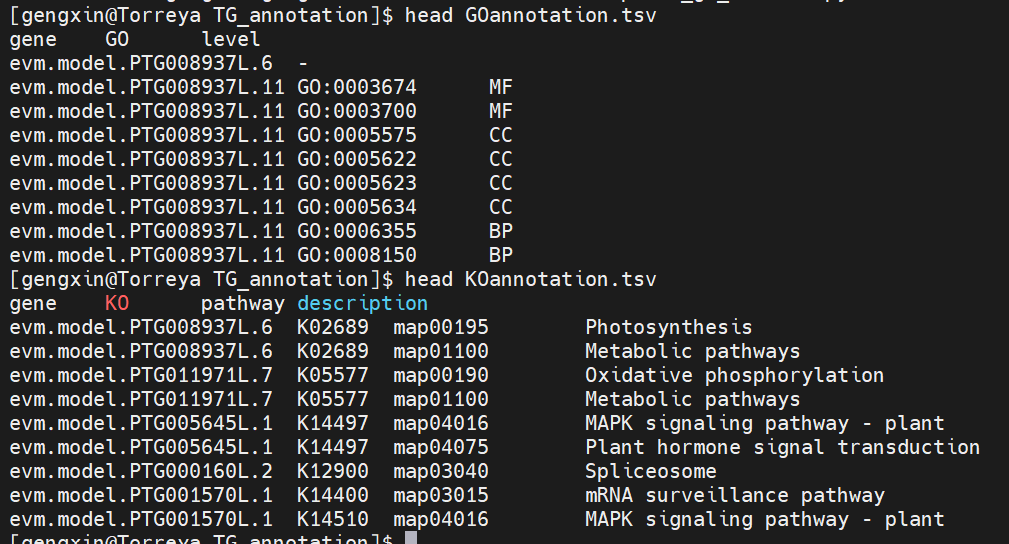
3、因为GOannotation.tsv没有description那一列,所以需要将GOannotation.tsv和go.tb双表关联。在【Rstudio】上输入代码run
library(tidyverse)
GO <- subset(GOannotation, select = -level)
t <- left_join(GO, go, by="GO")
write.table(t, file="E:/new_GOannotation1.tsv", sep = "\t", quote = FALSE, row.names = FALSE)根据提示安装各种依赖包,在右边的import dataset里输入go.tb文件和GOannotation.tsv文件。再run代码,生成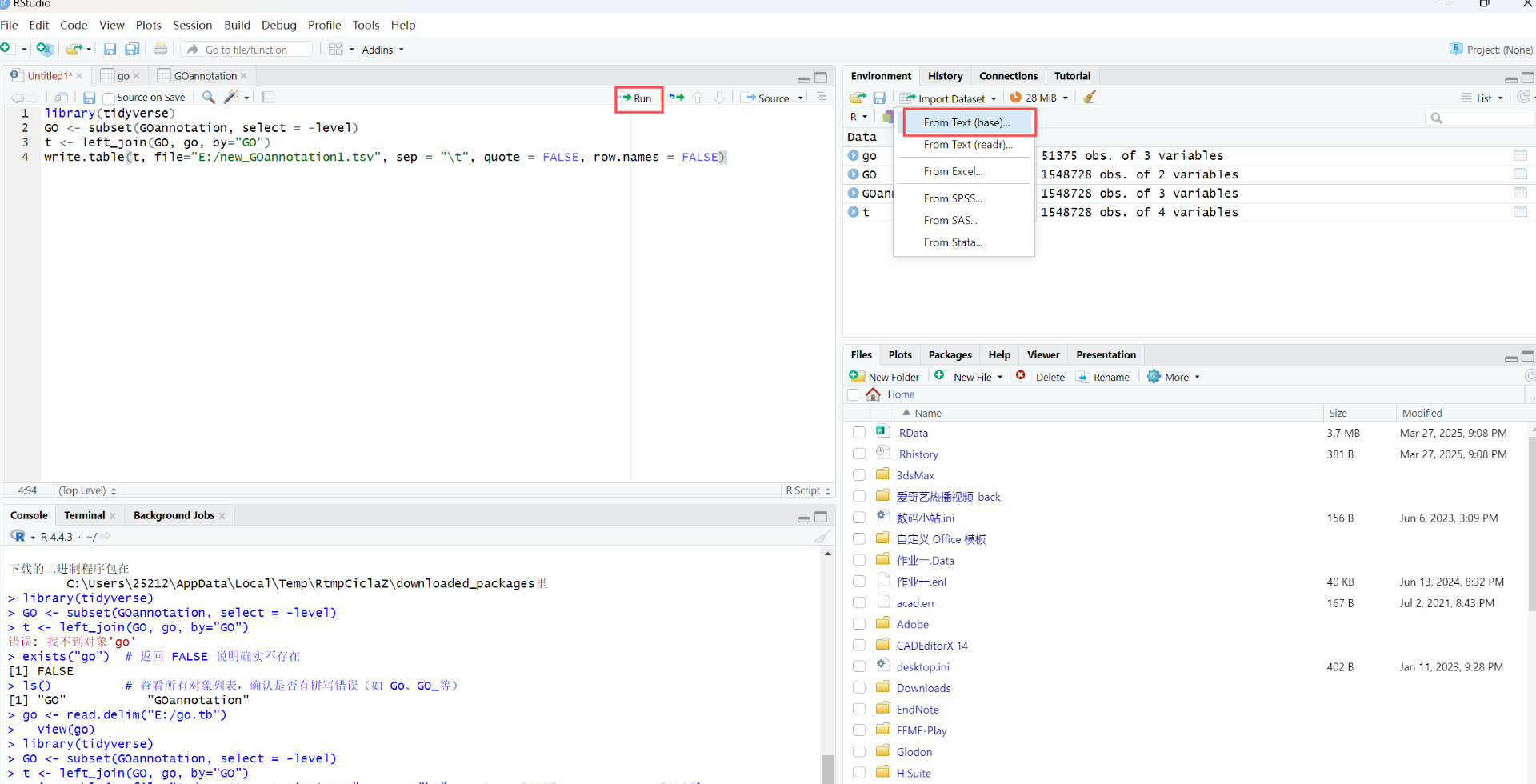
最后的GO注释文件