脚本目的
此脚本是用来是计算一些区间内是否偏好包含雌性或雄性偏向表达的基因,使用sys将区间文件和注释文件变成变量文件,优点是只需要在运行时修改注释文件和区间文件,不需要再脚本中修改路径
此脚本用法:
python analyze_regions_with_plot.py(此脚本) <GFF file> <Region file>
注意:
运行此脚本当前目录需要存在的文件:注释文件、区间文件、雄性偏好基因ID文件命名为male_biased_genes.csv、雌性偏好基因ID文件命名为female_biased_genes.csv
Region file示意图如下:
脚本
#!/usr/bin/env python3
# -*- coding: utf-8 -*-
"""
Integrated script: Region gene extraction + sex-biased gene intersection analysis +
Fisher's exact test (both female and male) + plotting with significance annotation
Usage:
python analyze_regions_with_plot.py <GFF file> <Region file>
"""
import pandas as pd
import sys
import os
from scipy.stats import fisher_exact
import matplotlib.pyplot as plt
import matplotlib as mpl
# === Font settings ===
try:
mpl.rcParams['font.family'] = 'Arial'
except:
mpl.rcParams['font.family'] = 'DejaVu Sans'
mpl.rcParams['pdf.fonttype'] = 42
mpl.rcParams['ps.fonttype'] = 42
# === Parameter check ===
if len(sys.argv) != 3:
print("Usage: analyze_regions_with_plot_v2.py <GFF file> <Region file>")
sys.exit(1)
gff_file = sys.argv[1]
region_file = sys.argv[2]
# === Create directory for intermediate results ===
output_dir = "region_results"
os.makedirs(output_dir, exist_ok=True)
# === Read region file ===
try:
regions_df = pd.read_csv(region_file, sep=r'\s+|,', header=None, engine='python')
except Exception as e:
print(f"❌ Failed to read region file: {e}")
sys.exit(1)
if regions_df.shape[1] < 3:
print("❌ Invalid file format error. Should contain 3 columns: <chr> <start> <end>")
sys.exit(1)
regions_df.columns = ['chr', 'start', 'end']
# === Read GFF file ===
print(f"📂 Reading GFF file: {gff_file}")
gff = pd.read_csv(gff_file, sep='\t', header=None, comment='#')
gff.columns = ['seqname', 'source', 'feature', 'start', 'end', 'score', 'strand', 'phase', 'attributes']
# === Extract all genes and parse gene_id ===
all_genes = gff[gff['feature'] == 'gene'].copy()
all_genes['gene_id'] = all_genes['attributes'].str.extract(r'ID=([^;]+)')
# === Load sex-biased gene data ===
try:
female_biased = pd.read_csv('female_biased_genes.csv')
male_biased = pd.read_csv('male_biased_genes.csv')
except FileNotFoundError as e:
print(f"❌ Sex-biased gene file not found: {e}")
sys.exit(1)
# === Output results file ===
output_file = 'region_intersection_results.txt'
with open(output_file, 'w') as f:
for idx, row in regions_df.iterrows():
chr_name, start_pos, end_pos = row['chr'], int(row['start']), int(row['end'])
# === Genes inside region ===
in_region = all_genes[
(all_genes['seqname'] == chr_name) &
(all_genes['start'] <= end_pos) &
(all_genes['end'] >= start_pos)
].copy()
in_file = os.path.join(output_dir, f'genes_in_{chr_name}_{start_pos}_{end_pos}.csv')
in_region[['gene_id']].to_csv(in_file, index=False)
f.write(f"✅ {chr_name}:{start_pos}-{end_pos} region extraction completed, {len(in_region)} genes total, saved as {in_file}\n")
# === Intersection with sex-biased genes ===
in_female = pd.merge(in_region[['gene_id']], female_biased, on='gene_id')
in_male = pd.merge(in_region[['gene_id']], male_biased, on='gene_id')
f.write(f" ♂♀ Intersection: female-biased {len(in_female)}, male-biased {len(in_male)}\n")
# === Genes outside region ===
out_region = all_genes.loc[~all_genes.index.isin(in_region.index)].copy()
out_female = pd.merge(out_region[['gene_id']], female_biased, on='gene_id')
out_male = pd.merge(out_region[['gene_id']], male_biased, on='gene_id')
f.write(f"📤 Total genes outside region: {len(out_region)}\n")
f.write(f" ♂♀ Outside region intersection: female-biased {len(out_female)}, male-biased {len(out_male)}\n")
# === Fisher test for female-biased ===
table_female = [
[len(in_female), len(in_region) - len(in_female)],
[len(out_female), len(out_region) - len(out_female)]
]
_, p_female = fisher_exact(table_female, alternative='two-sided')
# === Fisher test for male-biased ===
table_male = [
[len(in_male), len(in_region) - len(in_male)],
[len(out_male), len(out_region) - len(out_male)]
]
_, p_male = fisher_exact(table_male, alternative='two-sided')
# === Ratio & fold enrichment ===
female_ratio_in = len(in_female) / len(in_region) if len(in_region) > 0 else 0
female_ratio_out = len(out_female) / len(out_region) if len(out_region) > 0 else 0
male_ratio_in = len(in_male) / len(in_region) if len(in_region) > 0 else 0
male_ratio_out = len(out_male) / len(out_region) if len(out_region) > 0 else 0
female_fold = female_ratio_in / female_ratio_out if female_ratio_out > 0 else 0
male_fold = male_ratio_in / male_ratio_out if male_ratio_out > 0 else 0
# === Write results ===
f.write(f" 📊 Female-biased gene ratio (inside/outside): {female_ratio_in:.4f} / {female_ratio_out:.4f}\n")
f.write(f" 🎯 Female Fisher exact test P-value: {p_female:.6f}\n")
f.write(f" 📊 Female-biased gene enrichment fold: {female_fold:.4f}\n")
f.write(f" 📊 Male-biased gene ratio (inside/outside): {male_ratio_in:.4f} / {male_ratio_out:.4f}\n")
f.write(f" 🎯 Male Fisher exact test P-value: {p_male:.6f}\n")
f.write(f" 📊 Male-biased gene enrichment fold: {male_fold:.4f}\n\n")
print(f"✅ All analyses completed! Results saved to {output_file}")
# === Plotting section (with significance annotation) ===
print("🎨 Starting plotting with Fisher significance...")
regions, female_folds, male_folds, p_females, p_males = [], [], [], [], []
with open(output_file, 'r', encoding='utf-8', errors='ignore') as f:
lines = f.readlines()
current_region = None
for line in lines:
if line.startswith("✅"):
current_region = line.strip().split()[1]
elif "Female-biased gene enrichment fold" in line:
female_folds.append(float(line.strip().split(":")[-1]))
regions.append(current_region)
elif "Male-biased gene enrichment fold" in line:
male_folds.append(float(line.strip().split(":")[-1]))
elif "Female Fisher exact test P-value" in line:
p_females.append(float(line.strip().split(":")[-1]))
elif "Male Fisher exact test P-value" in line:
p_males.append(float(line.strip().split(":")[-1]))
# 确保长度一致
min_len = min(len(regions), len(female_folds), len(male_folds), len(p_females), len(p_males))
regions = regions[:min_len]
female_folds = female_folds[:min_len]
male_folds = male_folds[:min_len]
p_females = p_females[:min_len]
p_males = p_males[:min_len]
plt.figure(figsize=(12, 7))
plt.plot(regions, female_folds, label='Female-biased', color='orchid', marker='o',
linestyle='-', linewidth=2.5, markersize=8, markerfacecolor='white', markeredgewidth=2)
plt.plot(regions, male_folds, label='Male-biased', color='royalblue', marker='s',
linestyle='-', linewidth=2.5, markersize=8, markerfacecolor='white', markeredgewidth=2)
# === Add significance stars ===
def sig_star(p):
if p < 0.001: return '***'
elif p < 0.01: return '**'
elif p < 0.05: return '*'
else: return ''
for i, (pf, pm, yf, ym) in enumerate(zip(p_females, p_males, female_folds, male_folds)):
sig_f = sig_star(pf)
sig_m = sig_star(pm)
if sig_f:
plt.text(i, yf + 0.05, sig_f, ha='center', va='bottom', color='orchid', fontsize=12, fontweight='bold')
if sig_m:
plt.text(i, ym + 0.05, sig_m, ha='center', va='top', color='royalblue', fontsize=12, fontweight='bold')
# Baseline
plt.axhline(y=1.0, color='gray', linestyle='--', alpha=0.6, linewidth=1.5)
# Labels
plt.xlabel('Region', fontsize=14, fontweight='bold')
plt.ylabel('Enrichment Fold', fontsize=14, fontweight='bold')
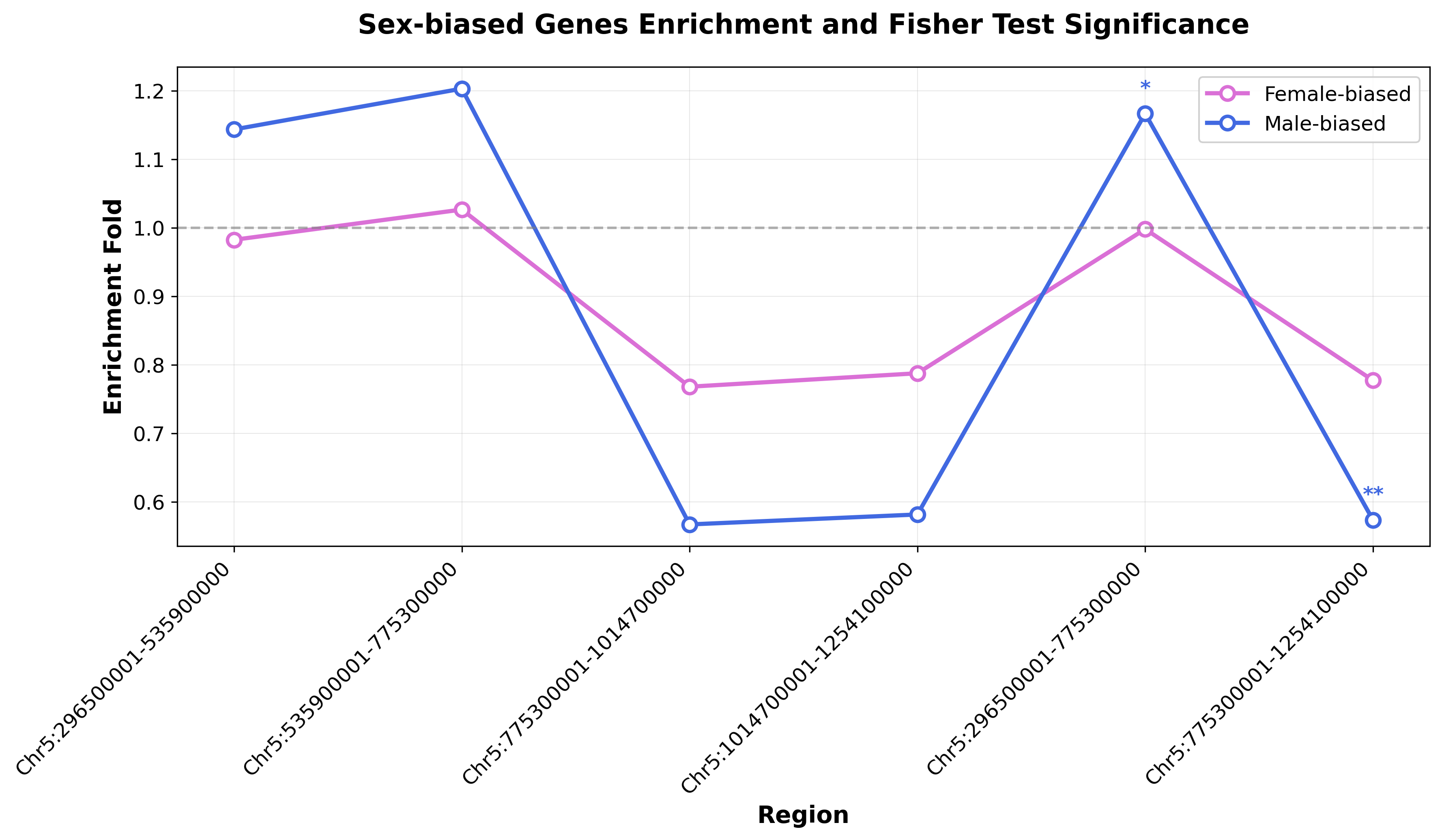
plt.title('Sex-biased Genes Enrichment and Fisher Test Significance',
fontsize=16, fontweight='bold', pad=20)
plt.xticks(rotation=45, ha='right', fontsize=12)
plt.yticks(fontsize=12)
plt.legend(fontsize=12, loc='best', framealpha=0.9)
plt.grid(True, alpha=0.3, linestyle='-', linewidth=0.5)
plt.tight_layout()
plt.savefig('gender_bias_enrichment_line.png', dpi=300, bbox_inches='tight')
plt.savefig('gender_bias_enrichment_line.pdf', dpi=300, bbox_inches='tight')
plt.show()
print("✅ Plotting completed! Output: gender_bias_enrichment_line.png/pdf")
生成文件
当前目录中会生成图片
region_results目录中会生成中间结果文件
【金山文档 | WPS云文档】 计算性偏好基因是否在性别决定区显著富集