开始进行数据比对,同时比对后的bam文件需要进行排序(SortSam)、标记duplication(MarkDuplicates)。
bwa mapping获得bam文件后,samtools进行格式转化。
# bwa-mem2 mem -R '@RG\tID:sample1\tLB:sample1\tSM:sample1\tPL:ILLUMINA' -t 5 R498.fasta sample1_1.filter.fq.gz sample1_2.filter.fq.gz |samtools view -Sb - > sample1.pe.bam # 获得bam文件 (base) root@961a4377e759:/home/Rmolle_calllsnp_work/Rmolle_results/bam_output_file# /home/software/bwa-mem2/bwa-mem2 mem -R '@RG\tID:QXG-1\tLB:QXG-1\tSM:QXG-1\tPL:DNBSEQ' -t 12 /home/Rmolle_calllsnp_work/Rmolle_genome_GCA025413875/Rmolle_genomic_GCA_025413875.1.fasta /home/Rmolle_calllsnp_work/Rmolle_results/fastp_results/QXG-1_R1.filter.fq.gz /home/Rmolle_calllsnp_work/Rmolle_results/fastp_results/QXG-1_R2.filter.fq.gz |samtools view -Sb - > QXG-1.pe.bam # 将ILLUMINA替换为DNBSEQ(替换前后结果文件相同) 注意:如果基因组文件的后缀为.fna时就会报错ERROR! Unable to open the file: 2.bwt.8bit.32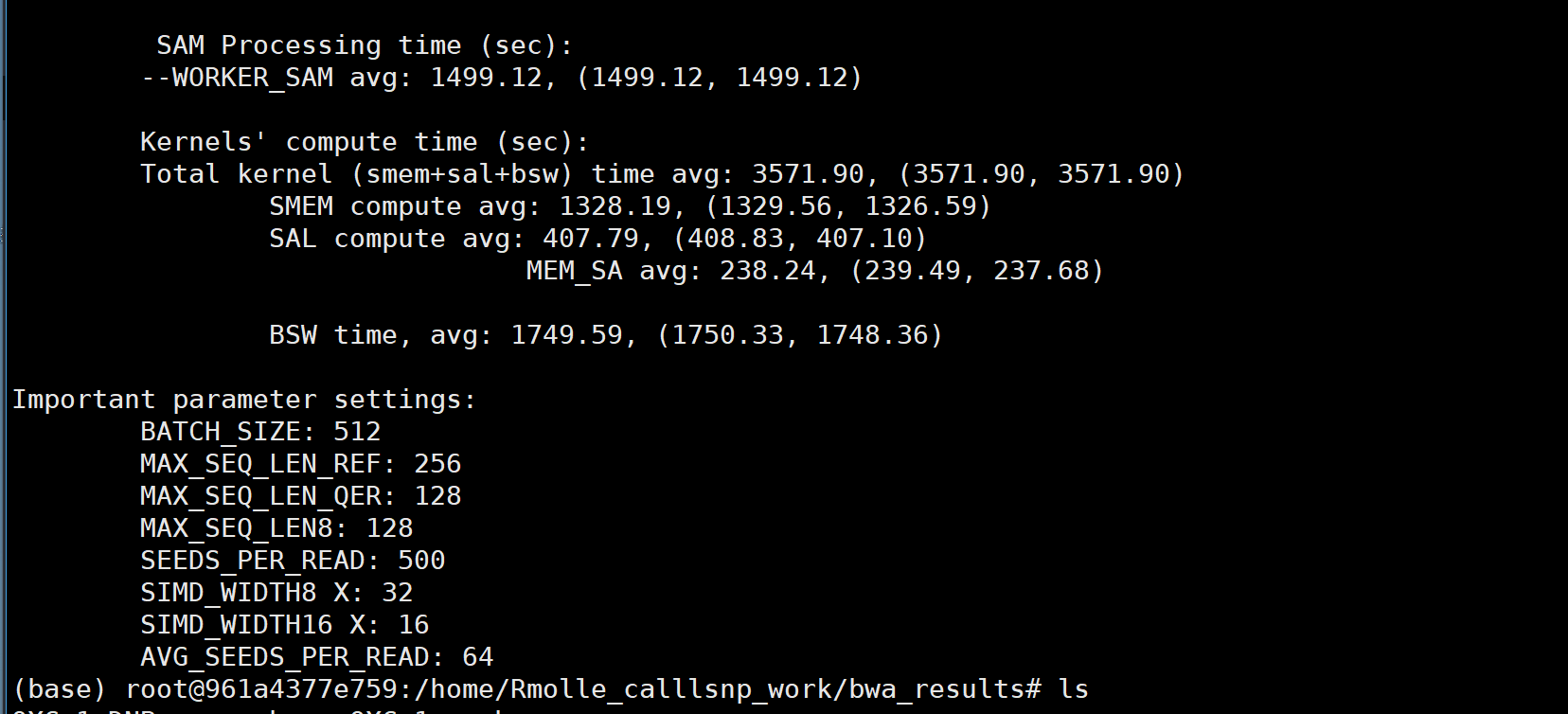
排序
# gatk SortSam --INPUT sample1.pe.bam --OUTPUT sample1.pe.sort.bam --SORT_ORDER coordinate --TMP_DIR ./tmp/sample1.tmp # 获得后缀sorted.bam的文件 (base) root@961a4377e759:/home/Rmolle_calllsnp_work/Rmolle_results/bam_sort_bygatk# /home/software/gatk-4.1.8.1/gatk SortSam --INPUT /home/Rmolle_calllsnp_work/Rmolle_results/bam_output_file/QXG-1.pe.bam --OUTPUT QXG-1.pe.sort.bam --SORT_ORDER coordinate --TMP_DIR ./tmp/QXG-1.tmp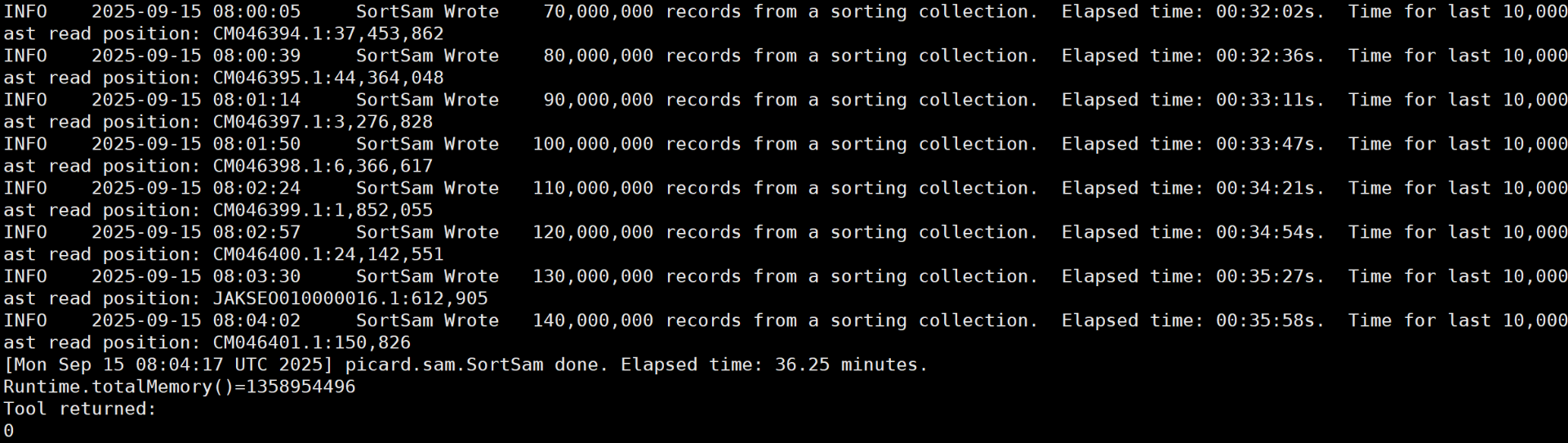
标记PCR重复
# gatk MarkDuplicates -I sample1.pe.sort.bam -O sample1.pe.sort.markdup.bam -M sample1.sort.markdup_metrics.txt # 获得文件后缀为sorted.markdup.bam、sorted.markdup_metrics.txt (base) root@961a4377e759:/home/Rmolle_calllsnp_work/Rmolle_results/bam_sort_bygatk# /home/software/gatk-4.1.8.1/gatk MarkDuplicates -I /home/Rmolle_calllsnp_work/Rmolle_results/bam_sort_markdup_bygatk/QXG-1.pe.sort.bam -O QXG-1.pe.sort.markdup.bam -M QXG-1.sort.markdup_metrics.txt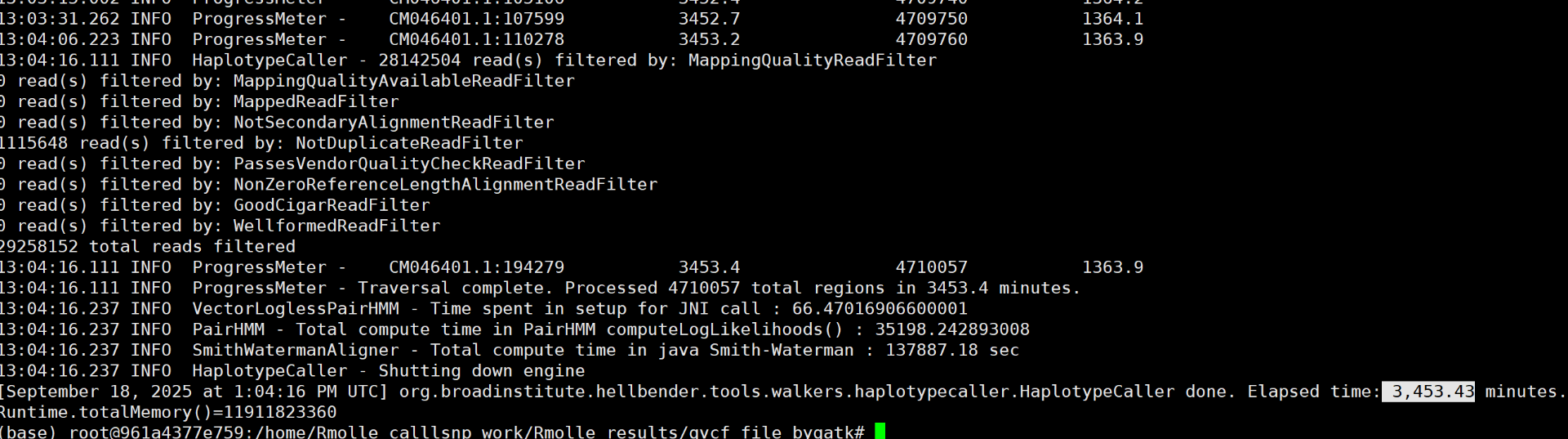
对获得的文件建立索引,不然得到的g.vcf文件和vcf文件没有内容
# samtools index sample1.pe.sort.markdup.bam (base) root@961a4377e759:/home/Rmolle_calllsnp_work/Rmolle_results# samtools index ./bam_sort_markdup_bygatk/QXG-1.pe.sort.markdup.bam