01 将分染色体的vcf文件合并成全基因组vcf文件
1. 首先,生成vcf列表文件:
ls INDEL.raw.chr*.vcf.gz > INDEX_raw_all_vcf.list
ls SNP.raw.chr*.vcf.gz > SNP_raw_all_vcf.list
# 如果有叶绿体等染色体,手动删除2. 使用gatk合并vcf文件
gatk \
--java-options "-Xmx10g -Djava.io.tmpdir=./tmp" \
MergeVcfs \
-I raw_vcf.list \
-O all.merge_raw.vcf02 过滤变异
gatk官网在2025年更新了Hard Filter的标准,选择最新的标准进行筛选:

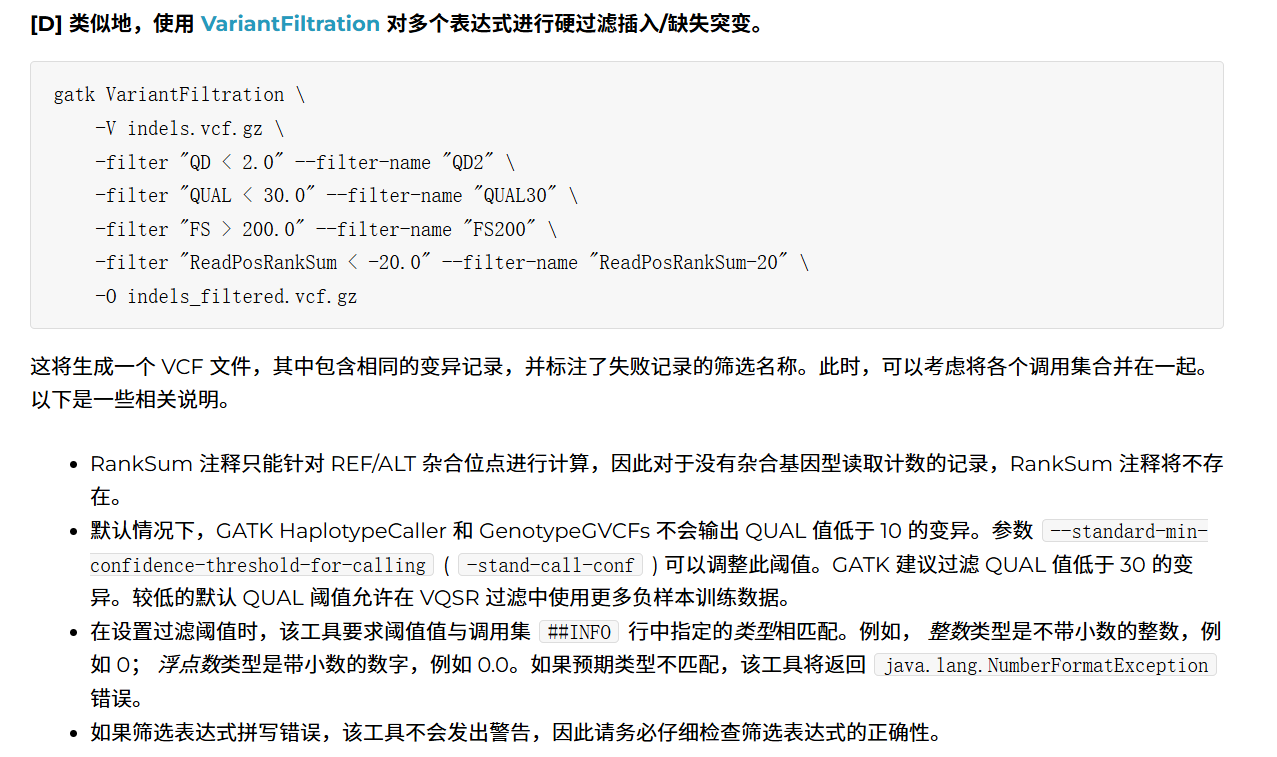
标记不合格的SNP和INDEL
# 标记不合格的SNP ## 过滤SNP(Filter列加标记) gatk VariantFiltration \ -V snps.vcf.gz \ -filter "QD < 2.0" --filter-name "QD2" \ -filter "QUAL < 30.0" --filter-name "QUAL30" \ -filter "SOR > 3.0" --filter-name "SOR3" \ -filter "FS > 60.0" --filter-name "FS60" \ -filter "MQ < 40.0" --filter-name "MQ40" \ -filter "MQRankSum < -12.5" --filter-name "MQRankSum-12.5" \ -filter "ReadPosRankSum < -8.0" --filter-name "ReadPosRankSum-8" \ -O snps_filtered.vcf.gz # 标记不合格的INDEL ## 过滤INDEL(Filter列加标记) gatk VariantFiltration \ -V indels.vcf.gz \ -filter "QD < 2.0" --filter-name "QD2" \ -filter "QUAL < 30.0" --filter-name "QUAL30" \ -filter "FS > 200.0" --filter-name "FS200" \ -filter "ReadPosRankSum < -20.0" --filter-name "ReadPosRankSum-20" \ -O indels_filtered.vcf.gz过滤不合格的SNP和INDEL
# 过滤不合格的SNP vcftools --gzvcf SNP.mark_lowq.chr1.vcf.gz --remove-filtered-all --min-alleles 2 --max-alleles 2 --recode --stdout |gzip -c >SNP.chr1.vcf.gz ## 但是在上述代码的基础上添加添加--recode-INFO-all,保留INFO信息 # 过滤不合格的INDEL vcftools --gzvcf indel.mark_lowq.chr1.vcf.gz --remove-filtered-all --min-alleles 2 --max-alleles 2 --recode --stdout |gzip -c >indel.chr1.vcf.gz变异位点数量统计
# 对比后,zcat更快 [root@3eb13a5fb4e6 7gvcf_snp_indel]$zcat genome.merge_raw.indel.vcf.gz | grep -v '^#' | wc -l (bcftools-1.22) [root@3eb13a5fb4e6 7gvcf_snp_indel]$bcftools view -H genome.merge_raw.snp.vcf.gz | wc -l