01 需要准备的文件
- gff文件(如果有的话,最好是gtf文件,gtf文件格式更严谨)
- 基因组文件
- vcf文件
02 利用annovar进行SNP注释
(1)准备基因组文件构建数据库
下载gffread:将gff文件转化为gtf文件
# 版本为为gffread v0.12.8 git clone https://github.com/gpertea/gffread cd gffread make release将gff文件转换为gtf文件
# gff转换为gtf gffread Rmolle_genomic_GCA_025413875.1.gff -T -o Rmolle.gtf将gtf文件转成refGene格式
# 利用conda下载gff3ToGenePred conda create -n ucsc -c bioconda ucsc-gtftogenepred conda activate ucsc # 转gtf成refGene格式 gtfToGenePred -genePredExt Rmolle.gtf Rmolle_refGene.txt提取序列文件
retrieve_seq_from_fasta.pl --format refGene --seqfile Rmolle_genomic_GCA_025413875.1.fasta -outfile Rmolle_refGeneMrna.fa Rmolle_refGene.tx
(2)SNP注释
- 参考:https://blog.csdn.net/qq_42756195/article/details/108545715?utm_medium=distribute.pc_relevant.none-task-blog-2~default~baidujs_baidulandingword~default-0-108545715-blog-139204318.235^v43^pc_blog_bottom_relevance_base8&spm=1001.2101.3001.4242.1&utm_relevant_index=3
- 参考:https://blog.csdn.net/qq_64400864/article/details/139204318
- 主要包含三种不同的注释方法:gene-based, region-based 和filter-based。基于基因的注释(Gene-based Annotation)揭示variant与已知基因直接的关系以及对其产生的功能性影响,基于区域的注释(Region-based Annotation)揭示variant 与不同基因组特定区域的关系,例如:它是否落在转录因子结合区域等,基于筛选的注释(Filter-based Annotation)则分析变异位点是否位于指定的数据库中,比如dbSNP, 1000G,ESP 6500等数据库。
- annovar 注释之前需要对文件格式进行转换,也可以使用vcf文件作为输入文件,只需要使用-vcfinput。
- table_annovar.pl和convert2annovar.pl都可以注释,并且table_annovar.pl可以一次性完成三种注释。但是自建库只能进行gene-based的注释。
- 需要注意的是,在使用convert2annovar.pl转换格式时,会有一个QC过滤的过程,所以建议使用该程序进行过滤和格式转换。
# 1.转换格式
## convert2annovar.pl转换格式
convert2annovar.pl -format vcf4 -allsample -withfreq all.clean.indel.vcf.gz > all.clean.indel.annovar.input
# 2.进行annovar注释
## 这部分内容为脚本测试
### 测试table_annovar.pl 和 annotate_variation.pl脚本有无区别(因为有看到教程说,如果是自己构建的数据库,则只有基于基因的注释类型能用;而table_annovar.pl一次性跑三个注释类型,也没有什么用)
### annotate_variation.pl脚本,--neargene 1000设置为1000,因为table_annovar.pl脚本默认的是1000
/home/software/annovar/annotate_variation.pl -geneanno --neargene 1000 -buildver Rmolle -dbtype refGene -outfile genome.indel1000.anno -exonsort genome.indel.vcf.annovar.input ./
### table_annovar.pl脚本
/home/software/annovar/table_annovar.pl genome.indel.vcf.annovar.input ./ -buildver Rmolle --outfile genome.indel1000.table.anno -remove -protocol refGene -operation g -nastring . -csvout
### 结果,两个任务跑出来的文件大小一致
## 选择annotate_variation.pl脚本进行注释,因为--neargene 可以设置为2000,范围更广
annotate_variation.pl -geneanno --neargene 2000 -buildver Rmolle -dbtype refGene -outfile genome.indel.anno -exonsort genome.indel.vcf.annovar.input ./
annotate_variation.pl -geneanno --neargene 2000 -buildver Rmolle -dbtype refGene -outfile genome.snp.anno -exonsort genome.snp.vcf.annovar.input ./03 结果统计
变异位点分布在基因组上的位置
less -NS XXX.variant_function | cut -f 1 | sort | uniq -c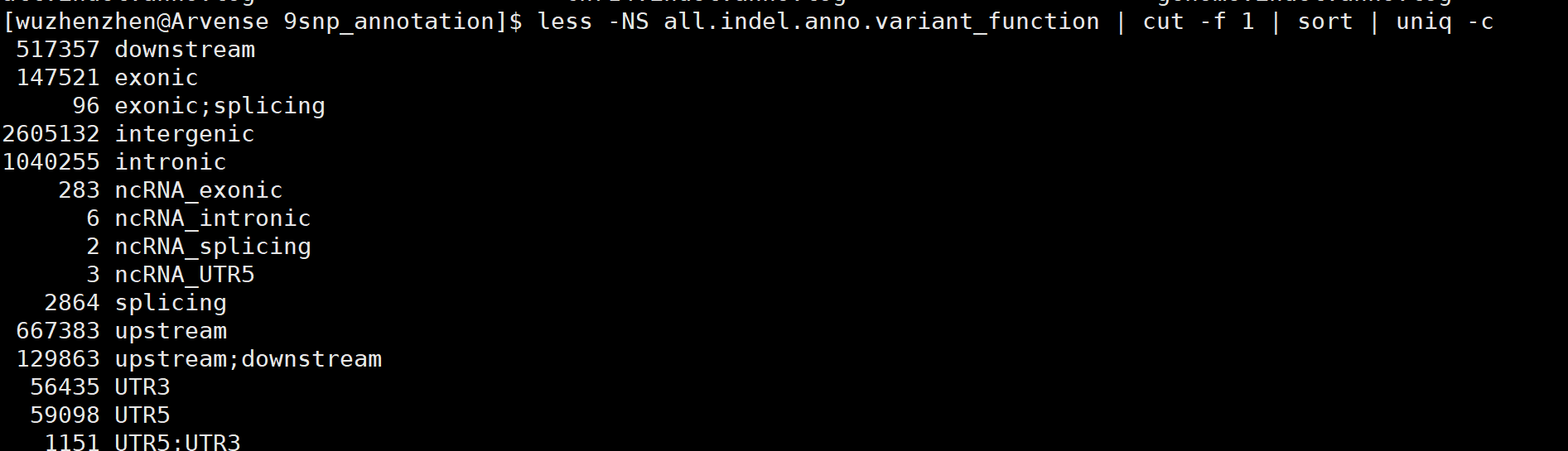
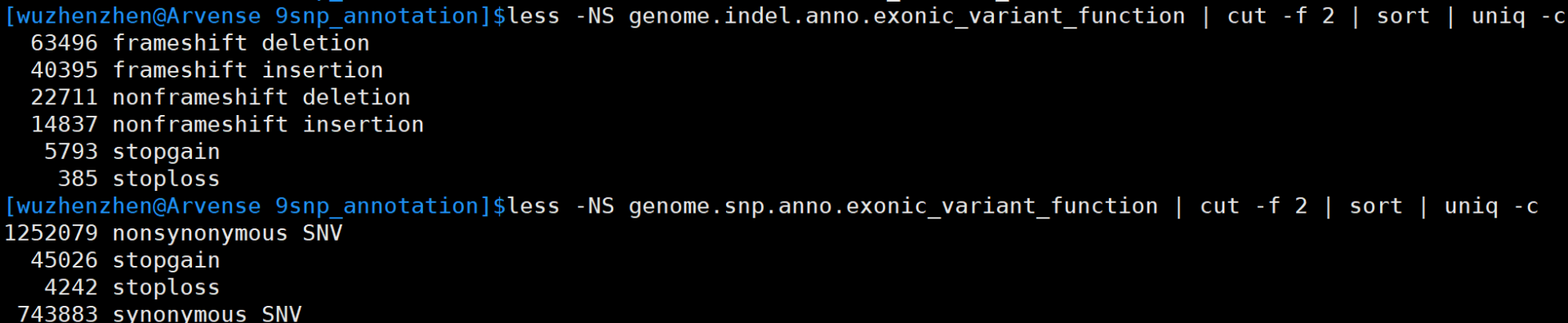
变异位点的类型
less -NS XXX.exonic_variant_function | cut -f 2 | sort | uniq -c