Description
侃大山,随便聊聊。有啥新奇特的Idea都可以分享。
侃大山,随便聊聊。有啥新奇特的Idea都可以分享。
分染色体合并:
# HaplotypeCaller最多可以设置4个线程,由于Java限制再增加也没有用
gatk --java-options "-Xmx10g -XX:ParallelGCThreads=4" HaplotypeCaller -R genome.fasta -I sample1.pe.sort.markdup.bam -ERC GVCF -O sample1.g.vcf.gz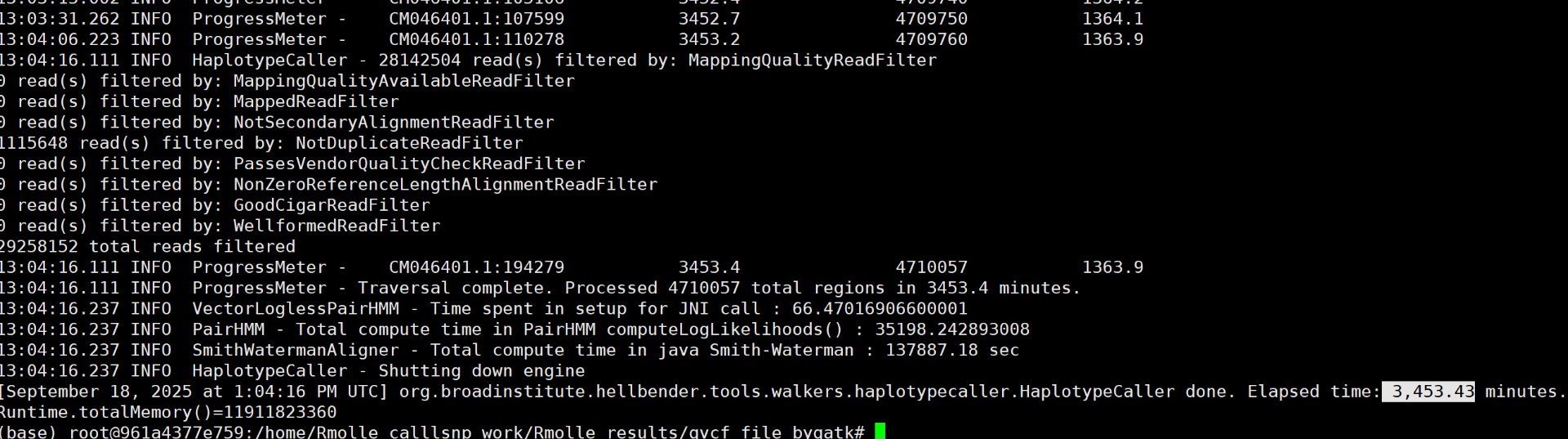
具体的报错信息:
在利用bwa mapping获得bam文件后,可以利用samtools对比对的结果进行统计。
# 统计每个样本的整体比对率(primary mapped)和双末端比对(properly paired)
samtools flagstat sample.bam
# 统计每个样本的覆盖情况(coverage)和平均测序深度(meandepth)
samtools coverage sample.bam
# 但要注意的是 coverage 参数在低版本的里面是没有的(如1.6),需要使用较新的版本的samtools,如1.21。问题:利用U盘从荧光定量 PCR仪器导出照片后,插到自己电脑上,U盘的所有内容都显示不出来。
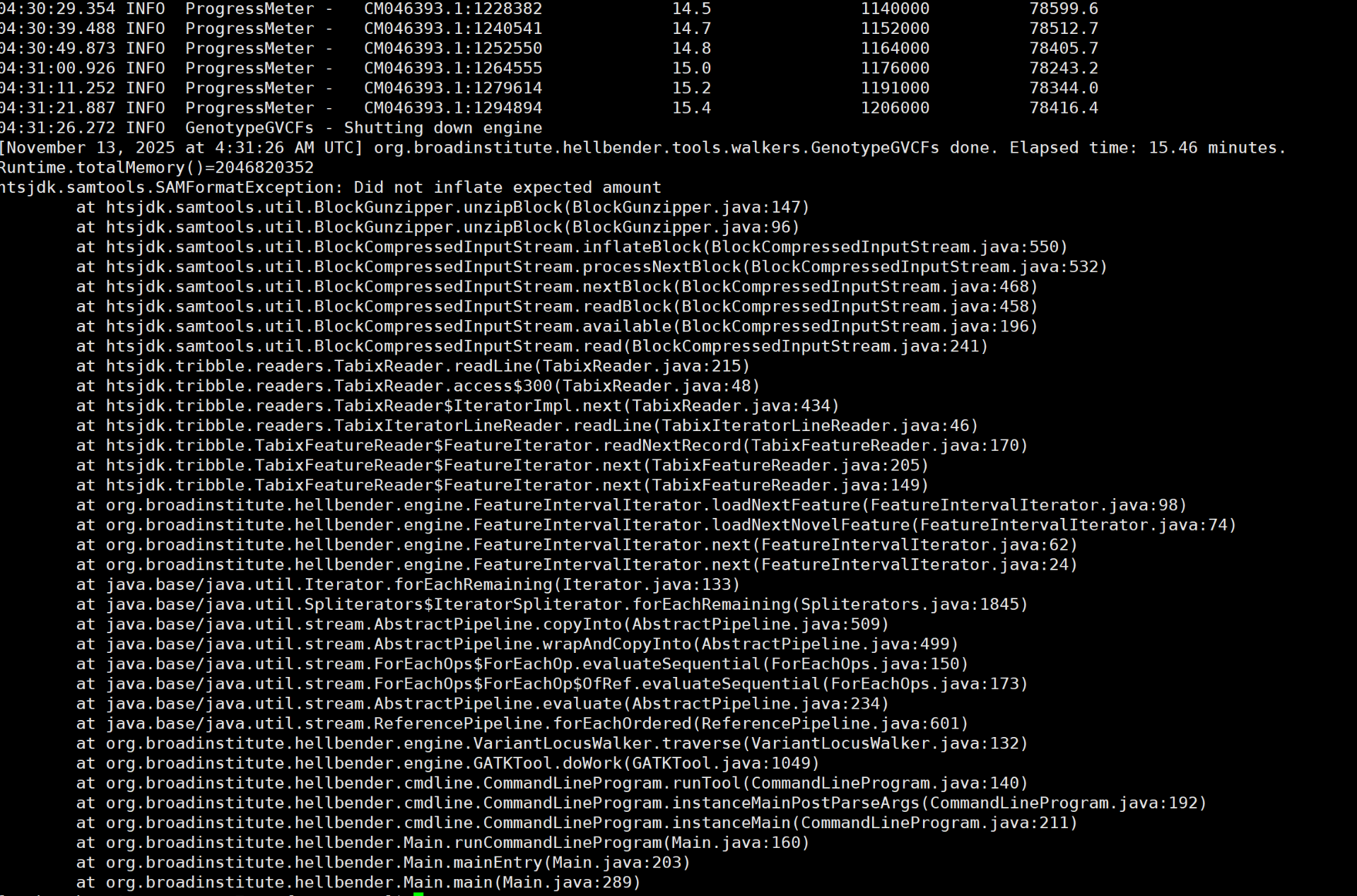
04:31:11.252 INFO ProgressMeter - CM046393.1:1279614 15.2 1191000 78344.0
04:31:21.887 INFO ProgressMeter - CM046393.1:1294894 15.4 1206000 78416.4
04:31:26.272 INFO GenotypeGVCFs - Shutting down engine
[November 13, 2025 at 4:31:26 AM UTC] org.broadinstitute.hellbender.tools.walkers.Gen由于开题老师提出应关注一下我课题中研究的基因在拟南芥中的功能作用,因此进行归纳总结。
一、数据的准备