Description
侃大山,随便聊聊。有啥新奇特的Idea都可以分享。
侃大山,随便聊聊。有啥新奇特的Idea都可以分享。
前期准备:论坛 考种:excle表格制作教程
前期准备:论坛 考种:excle表格制作教程
本教程主要功能是基于种质资源数据制作EXCLE表。
基于分区,填写区域颜色编号、基地名称编号、区域颜色、小区域编号、行和列(行和列必须为00的格式)(小区域如果超过9,则这个编号规则不太适用);
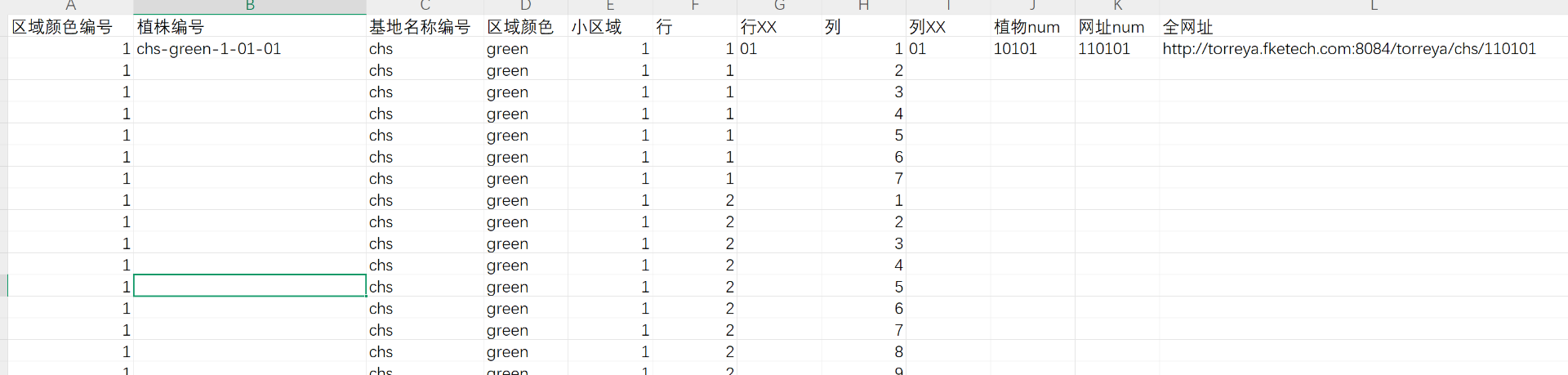
按照其他信息,生成植株编号、植物num和全网址。
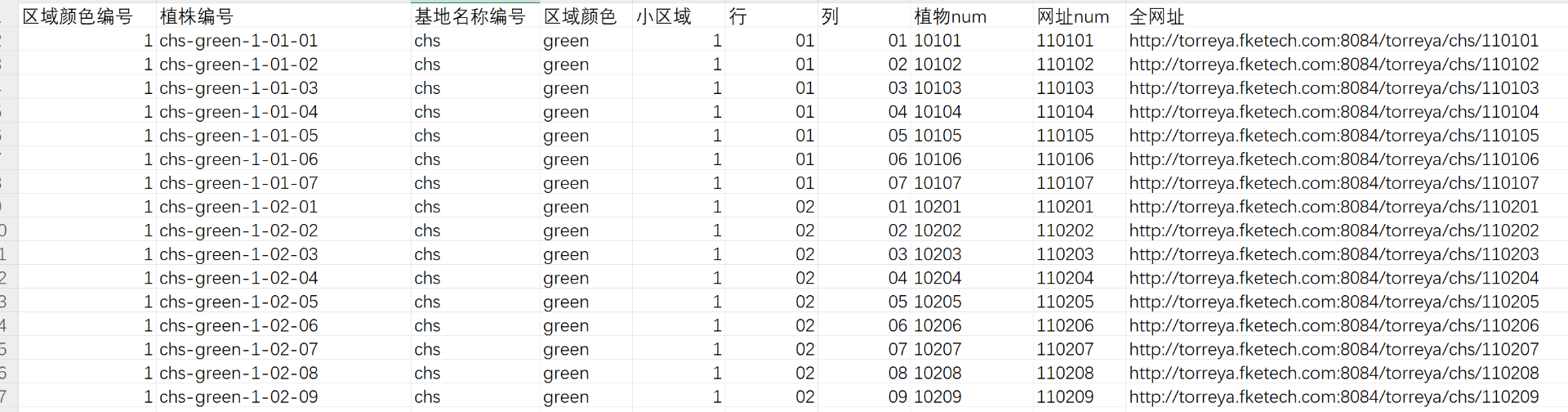
将整个excle表以纯文本粘贴。
雅各布散度(Jensen–Shannon Divergence, JSD)
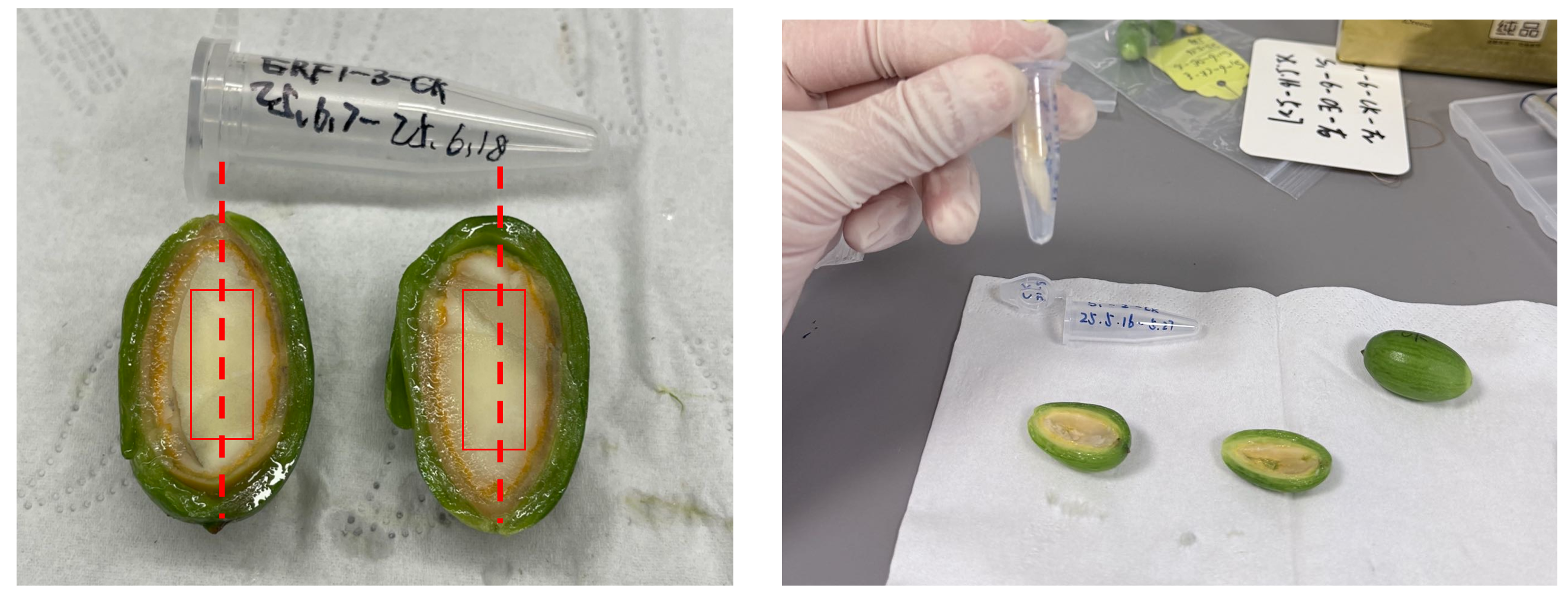 大孢子的种仁取样时是纵向取样的,因此在制作切片时应该将样品朝向一致摆放,沿虚线位置纵切
大孢子的种仁取样时是纵向取样的,因此在制作切片时应该将样品朝向一致摆放,沿虚线位置纵切
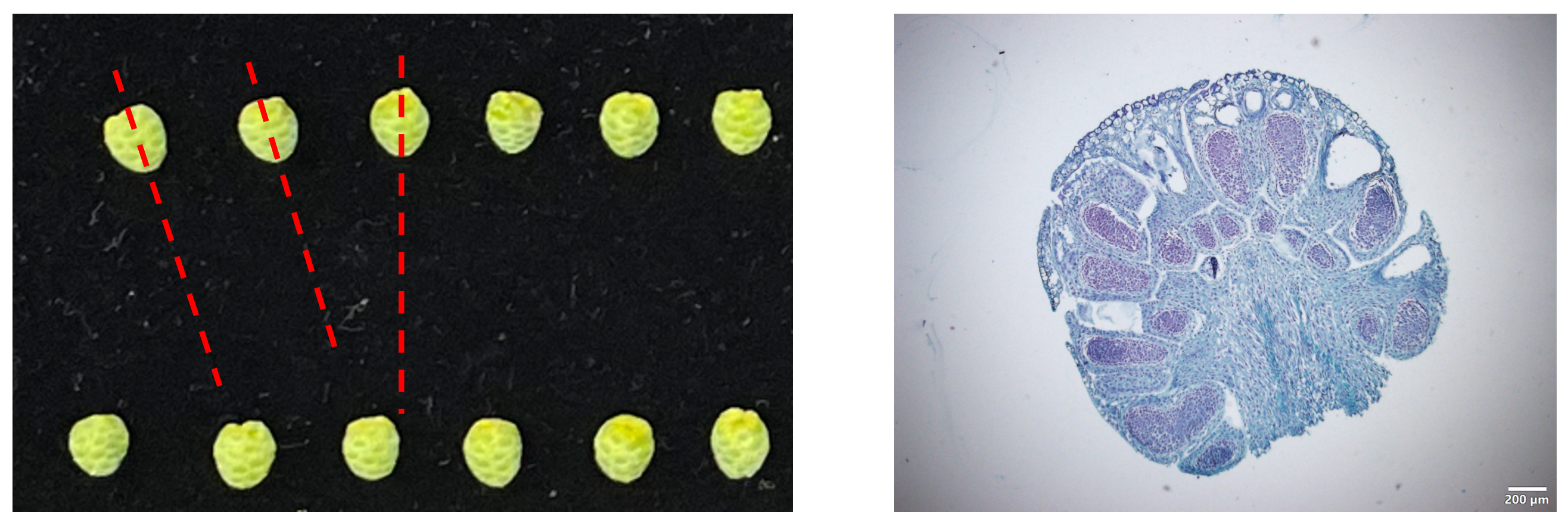 小孢子为图中所示,制作石蜡切片应使得其沿虚线位置纵切,使其在显微镜下呈现右图的效果
小孢子为图中所示,制作石蜡切片应使得其沿虚线位置纵切,使其在显微镜下呈现右图的效果
原始数据在/data/public_92/Torreya_grandis/NCBI-20230420,是鑫哥整理的版本
容器用的是鑫哥的061b51fa0bb3
数据处理
把从ncbi上下载的sra数据转换为fastq
下载sratoolkit
1. 创建专用环境
conda create -n sra_tools python=3.9
2. 激活环境
conda activate sra_tools
3. 安装 sratoolkit
conda install -c bioconda sra-tools
使用
单端测序(或RNA测序数据,最开始受网上信息的误导,博主讲RNA数据用单端测序,但是运行完发现,下载数据是双端测序的数据):
fastq-dump SRR23105320.sra -O /home/lry
转换完成后,根据华东师兄的方法,压缩为gz文件
双端测序
fastq-dump SRR14306907.sra --split-3 --gzip -O ./
用华东师兄的流程跑,config,info文件的内容
PROJECT: ganhan
董老师发我的娄老师那边的雌雄干旱转录组的fpkm,但是是孙老师注释的版本,需要先转换一下
放在92: /data3/liruiyuan/blast/home/work/positive/biada/cixiongganhan_fpkm.csv
从鹏哥那里来的sun-ncc对应表
用下面这个代码替换的。是sun到ncc版本的替换,适用于sun到其他版本。
ncc到sun版本不一定适用。
def replace_gene_ids(expression_file, id_map_file, output_file):
id_map = {}
with open(id_map_file, 'r') as f:
for line in f:
line = line.strip()
if not line or '\t' not in line:
continue
parts = line.split('\t')
if len(parts) == 2: