Description
如果有什么常用的资源,可以大家一起分享的,请在此处分享。
如果有什么常用的资源,可以大家一起分享的,请在此处分享。
【金山文档 | WPS云文档】 利用Guetzli图像压缩技术优化农业无人机图像存储与传输 https://www.kdocs.cn/l/clLjhwsLIPap
金山文档链接:
【金山文档 | WPS云文档】 通过IGV查询染色体臂易位断点
https://www.kdocs.cn/l/cbupgU1LTOfU
以下是全文内容:
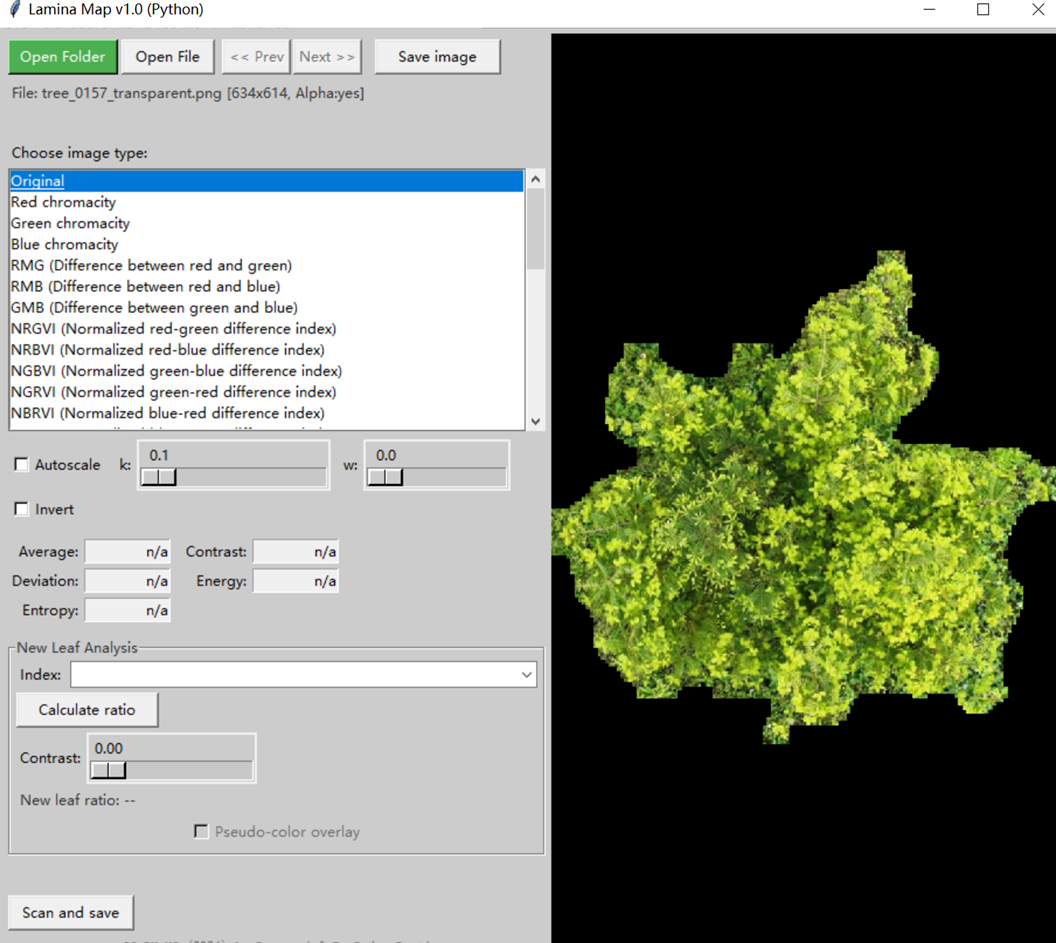
【金山文档 | WPS云文档】 Leaflaminamap导出指数图名称对应关系 https://www.kdocs.cn/l/ce236FiiRRBO
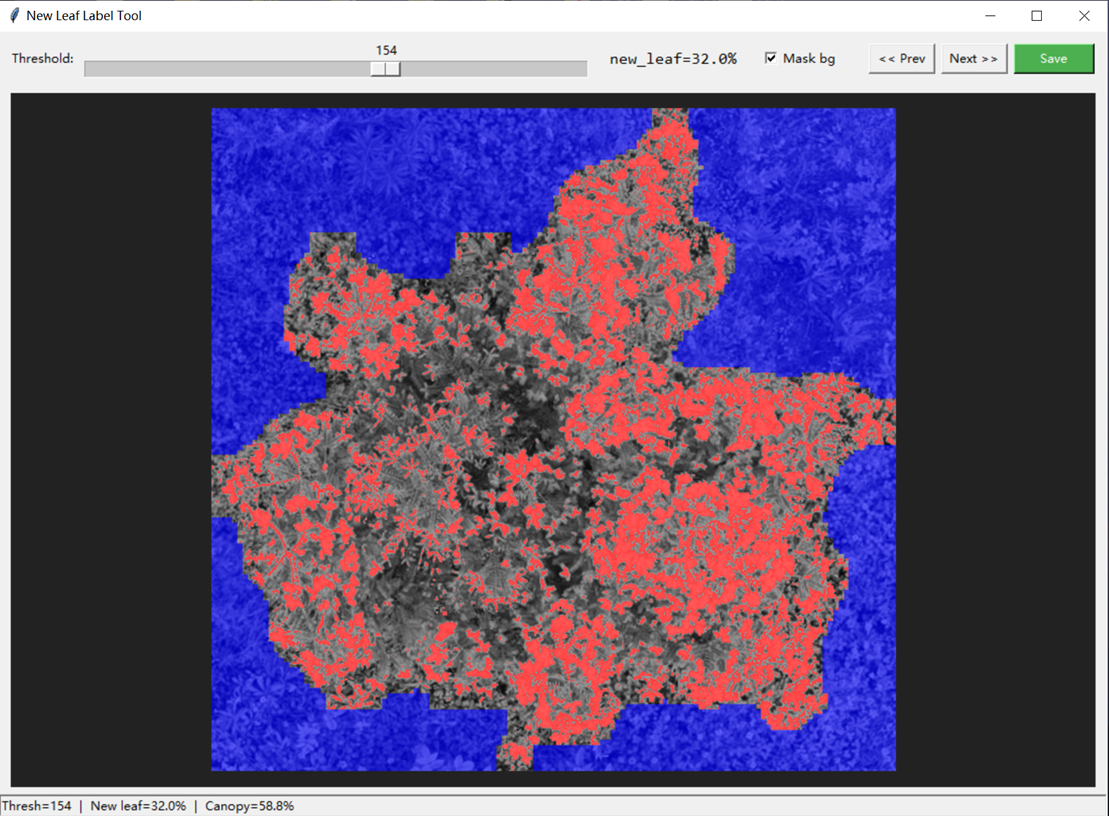
【金山文档 | WPS云文档】 LeaflaminaMap改进版 https://www.kdocs.cn/l/ccABDSwAjlRw